
The Hungate 1000 Project was a massive undertaking: namely, sequencing the genome of 1000 microorganisms cultured from ruminant animals all over the world, and was both coordinated and led by the Rumen Microbial Genomics Network. After years of hard work by some incredible researchers, the Hungate 1000 has just been published in the Nature Biotechnology Journal!
The Hungate 1000 Project was named in honor of Dr. Robert Hungate, whose works on rumen microorganisms from the 1930s through the 1960s revolutionized the field and is the basis for much of the work on ruminants today. The goal of the Project was to sequence the genomes of 1000 microorganisms cultured from the rumen of different host species. The vast majority of rumen microorganisms are not cultivable, largely because they require very particular growing conditions. By sequencing genomes of cultivable isolates, the Project not only provides a more comprehensive database of genomic information, but will provide insight into the life of these microorganisms. The full catalogue of isolates which were donated to the Hungate 1000 Project can be found here: I was able to donate isolates which I cultured from North American moose (Alces Alces) from Vermont.
The RMG Network also ran the Global Rumen Census Project. The GRC Project involved sequencing hundreds of samples from ruminants all over the globe, to study trends in diet, host, and geographic location on a massive scale. I was able to contribute to this project by donating moose and other ruminant samples.
Additional discussion on this achievement can be found in posts from the Joint Genome Institute (JGI) and the New Zealand Agricultural Greenhouse Gas Research Centre (NZAGRC). From the NZAGRC post, “The Hungate1000 was funded by the New Zealand Government through the Ministry for Primary Industries in support of the Livestock Research Group of the Global Research Alliance on Agricultural Greenhouse Gases (GRA), which is administered by the NZAGRC. The genome sequencing and analysis component of the project was supported by the United States Department of Energy’s Joint Genome Institute (JGI), via its Community Science Program.”
Cultivation and sequencing of rumen microbiome members from the Hungate1000 Collection
Rekha Seshadri, Sinead C Leahy, Graeme T Attwood, Koon Hoong Teh, Suzanne C Lambie, Adrian L Cookson, Emiley A Eloe-Fadrosh, Georgios A Pavlopoulos, Michalis Hadjithomas, Neha J Varghese, David Paez-Espino, Hungate1000 project collaborators, Rechelle Perry, Gemma Henderson, Christopher J Creevey, Nicolas Terrapon, Pascal Lapebie, Elodie Drula, Vincent Lombard, Edward Rubin, Nikos C Kyrpides, Bernard Henrissat, Tanja Woyke, Natalia N Ivanova & William J Kelly
Abstract
Productivity of ruminant livestock depends on the rumen microbiota, which ferment indigestible plant polysaccharides into nutrients used for growth. Understanding the functions carried out by the rumen microbiota is important for reducing greenhouse gas production by ruminants and for developing biofuels from lignocellulose. We present 410 cultured bacteria and archaea, together with their reference genomes, representing every cultivated rumen-associated archaeal and bacterial family. We evaluate polysaccharide degradation, short-chain fatty acid production and methanogenesis pathways, and assign specific taxa to functions. A total of 336 organisms were present in available rumen metagenomic data sets, and 134 were present in human gut microbiome data sets. Comparison with the human microbiome revealed rumen-specific enrichment for genes encoding de novosynthesis of vitamin B12, ongoing evolution by gene loss and potential vertical inheritance of the rumen microbiome based on underrepresentation of markers of environmental stress. We estimate that our Hungate genome resource represents ∼75% of the genus-level bacterial and archaeal taxa present in the rumen.
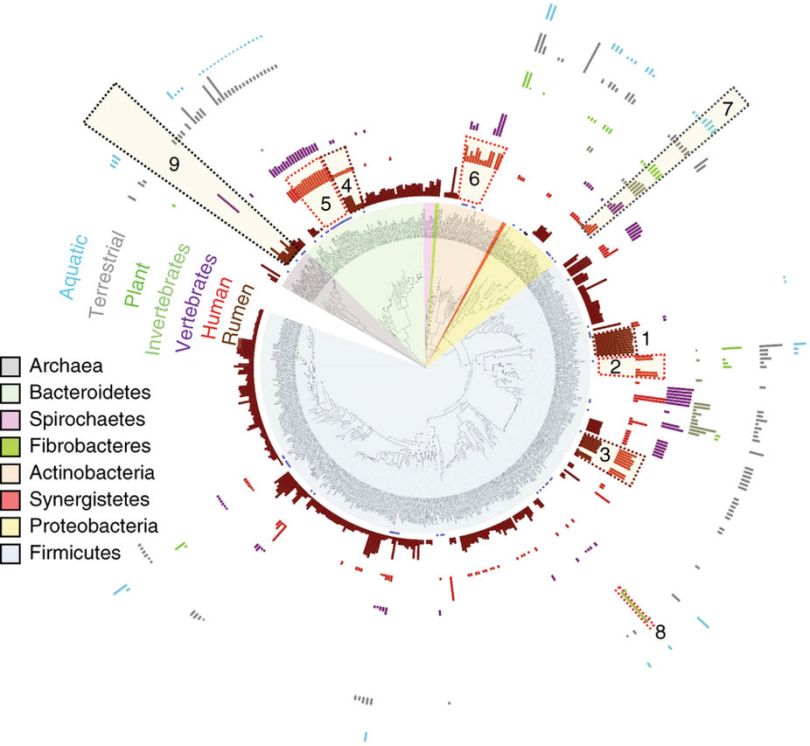
Featured Image; https://www.nature.com/articles/nbt.4110/figures/4
Figure 4 Maximum likelihood tree based on 16S rDNA gene alignment of rumen strains. The tree clades are color coded according to phylum. Multi-bar-chart depicting the average % coverage of total CDS of an isolate by metagenome samples from each ecosystem category was drawn using iTOL55. Dashed boxes highlight interesting examples of recruitment such as isolates detected in both rumen and human samples (maroon boxes) or detected in human but not rumen samples (red boxes), and others. Number key is as follows (average % coverage is given in parentheses): 1. Sharpea azabuensis str. (∼88%), Kandleria vitulina str. (∼87%); 2. Staphylococcus epidermidis str. (∼40%), Lactobacillus ruminis str. (∼51%); 3. Streptococcus equinus str. (∼38% by rumen, ∼35% by human); 4. Prevotella bryantii str. (∼38% by rumen, ∼9% by human); 5. Bacteroides spp.(∼38%); 6. Bifidobacterium spp. (∼24%), Propionibacterium acnes (∼39%); 7. Shigella sonnei (∼30% by human), E. coli PA3 (∼31% by human), Citrobacter sp. NLAE-zl-C269 (20% by human); 8. Clostridium beijerinckii HUN142 (87% by plant); 9. Methanobrevibacter spp. (∼32%⋆Figure 4 Maximum likelihood tree based on 16S rDNA gene alignment of rumen strains. The tree clades are color coded according to phylum. Multi-bar-chart depicting the average % coverage of total CDS of an isolate by metagenome samples from each ecosystem category was drawn using iTOL55. Dashed boxes highlight interesting examples of recruitment such as isolates detected in both rumen and human samples (maroon boxes) or detected in human but not rumen samples (red boxes), and others. Number key is as follows (average % coverage is given in parentheses): 1. Sharpea azabuensis str. (~88%), Kandleria vitulina str. (~87%); 2. Staphylococcus epidermidis str. (~40%), Lactobacillus ruminis str. (~51%); 3. Streptococcus equinus str. (~38% by rumen, ~35% by human); 4. Prevotella bryantii str. (~38% by rumen, ~9% by human); 5. Bacteroides spp.(~38%); 6. Bifidobacterium spp. (~24%), Propionibacterium acnes (~39%); 7. Shigella sonnei (~30% by human), E. coli PA3 (~31% by human), Citrobacter sp. NLAE-zl-C269 (20% by human); 8. Clostridium beijerinckii HUN142 (87% by plant); 9. Methanobrevibacter spp. (~32%). The innermost circle identifies Hungate isolates of fecal (★) or salivary (♦) origin. Please refer to Supplementary Table 9 for data and other specifics.
