I'm an assistant professor of animal and veterinary studies at the University of Maine, Orono, studying how animals get their microbes. I am also the Founder and Lead of the Microbes and Social Equity working group.
The Maine Farmcast, a podcast led by my friends and colleagues at UMaine Cooperative Extension, Drs. Colt Knight and Glenda Pereira, invited me to chat about my research and teaching recently. Check out Episode 111: ‘Gut Check, Microbiomes in Agriculture and Health with Dr. Sue Ishaq’! I had a lot of fun catching up with Colt and sharing horror stories about messy research on the farm and in the lab.
Maine Farmcast
“The Maine Farmcast features weekly conversations with experts from across the country sharing insights and advice for both new and seasoned farmers running operations of any size. Hosted by three livestock specialists from the University of Maine Cooperative Extension, the show brings you world-class expertise paired with practical advice about how to apply cutting-edge research to improve the efficiency and sustainability of your farming operation.”
Events will be hosted January – December in 2026, on the last Wednesday of every month, 11:00 – 13:00 pm ET. Presented over Zoom.
After each talk, we will continue the discussions in an informal social meeting with MSE. All speakers and members of the audience are welcome to join the social meeting.
New this year: the live session will be available free, but the on-demand video-recording will only be available to MSE members for the first year (and available to the public afterwards).
Summary:
Microorganisms are critical to many aspects of biological life, including human health. The human body is a veritable universe for microorganisms: some pass through but once, some are frequent tourists, and some spend their entire existence in the confines of our body tissues. The collective microbial community, our microbiome, can be impacted by the details of our lifestyle, including diet, hygiene, health status, and more, but many are driven by social, economic we, medical, or political constraints that restrict available choices that may impact our health. Access to resources is the basis for creating and resolving social equity—access to healthcare, healthy foods, a suitable living environment, and to beneficial microorganisms, but also access to personal and occupational protection to avoid exposure to infectious disease. This speaker series explores the way that microbes connect public policy, social disparities, and human health, as well as the ongoing research, education, policy, and innovation in this field.
Dr. Sarah Elton is an Assistant Professor and Eakin Chair in Critical Qualitative Health Research Methodology at the University of Toronto’s Dalla Lana School of Public Health. She researches at the nexus of food systems, ecosystems and human health, considering the more-than-humans who co-produce health, including microbes. In 2021, she was the first qualitative researcher to be recognized by the Gairdner Foundation when she won a Gairdner Early Career Investigator Award. Previous to her academic career, Sarah worked as a journalist and is the author of two Canadian bestselling nonfiction books, Locavore and Consumed: Food for a Finite Planet. In this presentation, she draws on more-than-human methodologies and her own field work in food systems to explore different ways social scientists can conduct research with microbes. She focuses on how critical qualitative research methodologies can enable scholars to investigate the social and political forces that shape human-microbe relations.
Whether we are interested in the way coastal wetlands sequester carbon, how diet affects us differently, or how public and environmental health are inextricably linked – research on the microbiome can reveal how systems connect. Join us for two days to learn about how microbial communities impact ecosystems, food production, health, and more; to hear from experts researching these issues in New England and beyond; and discuss the technology and data analysis which can boost your own research.
UMaine Buchanan Alumni House, 160 College Ave, Orono, ME 04473
Dr. Sue Ishaq, PhD, Associate Professor of Microbiomes, Associate Director of Special Projects in the School of Food and Agriculture at the UMaine; member of the Board of Directors at ASM; Founder and Lead of the Microbes and Social Equity working group.
9:15 – 10:15 Plenary Presentation
“The second messenger c-di-GMP in Bordetella”
Dr. Federico Sisti, Ph.D., Investigator, Institute of Biotechnology and Molecular Biology (IBBM) of the National Scientific and Technical Research Council (CONICET). Dr. Sisti is incoming President-Elect at the American Society of Microbiology, and the first ASM President representing its global membership.
10:15 – 10:30 Break
10:30 – 12:00 Session 1: Microbial mayhem or tiny partners in health?
“Leveraging organ-on-chip technology to study gut microbiome effects on human health and disease“, Dr. Dani Brassino, PhD, Assistant Professor of Gut Microbiome and Cancer Interactions, The University of Vermont College of Medicine.
Following a B.S. in Nutrition Science and at the University of Texas, Dani Brasino acquired a PhD in Chemical Engineering at the University of Colorado. At CU, she developed synthetic phospholipids as part of an artificial cell project and acquired experience in microfluidics fabrication. Combining these experiences, she led development of a novel polycarbonate-based organ-on-chip platform as a postdoc at Oregon Health and Science University. Now her lab, the μMicrobiome Lab, aims to further develop and apply gut microbiome-on-chips to study the relationship between the human gut microbiome, distal disease progression, and therapeutic efficacy.
“Genetic and Microbiome Control of Addiction-like Behaviors in Mice”, Dr. Jason Bubier, Ph.D., Senior Research Scientist, Center for Addiction Biology, The Jackson Laboratory
Jason A. Bubier, Ph.D., is a Senior Research Scientist at The Jackson Laboratory, where he leads research at the intersection of genetics, behavior, and addiction biology. His work focuses on uncovering the genetic and physiological factors that shape vulnerability to addictive substances, with current projects examining the microbiome’s influence on behavior, the genetic architecture of opioid‑induced respiratory depression, and systems‑level pathways involved in addiction.
Dr. Bubier has spent more than two decades at The Jackson Laboratory, advancing through multiple research roles and contributing to major initiatives in functional genomics and behavioral genetics. He has authored more than 80 scientific publications, and his work is widely cited across fields including immunology, bioinformatics, and addiction science.
“Food insecurity, gut microbiome, health”, Dr. Maria Carlota Dao, Ph.D., Assistant Professor of Human Nutrition, University of New Hampshire.
Dr. Maria Carlota Dao is an Assistant Professor of Human Nutrition in the Department of Agriculture, Nutrition, and Food Systems at the University of New Hampshire. As an interdisciplinary scientist focused on obesity research, she investigates the interplay of cardiometabolic risks, dietary and psychosocial factors, and the gut microbiota. Through this work she seeks to address obesity in health disparity populations. Prior to becoming a faculty member at UNH, Dr. Dao worked as a scientist at the Jean Mayer USDA Human Nutrition Research Center on Aging at Tufts University. Her training includes a postdoctoral fellowship at the Sorbonne University and INSERM (Institute of Cardiometabolism and Nutrition) in France, and a PhD in Biochemical and Molecular Nutrition from the Friedman School of Nutrition Science and Policy at Tufts University.
“Gut Anthro“, Dr. Amber Benezra, Assistant Professor of Science and Technology Studies at Stevens Institute of Technology
Dr. Benezra is a sociocultural anthropologist researching how studies of the human microbiome intersect with biomedical ethics, public health/technological infrastructures, and care. In partnership with human microbial ecologists, she has been developing an “anthropology of microbes” to address global health problems across disciplines. Her book, Gut Anthro (published by University of Minnesota Press in 2023) is the first ethnography of the microbiome.
“One Codex in Action“, Christopher Smith, One Codex.
12:00 – 1:15 Lunch, provided
1:15 – 2:45 Session 2: Biochemical geniuses: microbes from soil to sea
“How to build a microbiome to “breed” better plant-inoculants.” Dr. Anna O’Brien, PhD., Assistant Professor of Molecular, Cellular, and Biomedical Sciences, University of New Hampshire.
She is originally from the Pacific Northwest. She received her B.S. in Plant Biology from the University of Washington, her PhD from the University of California Davis, where she worked with Jeffrey Ross-Ibarra and Sharon Strauss on the impacts of rhizosphere biota on trait divergence and adaptation in the wild relatives of maize. Anna Post-doc’ed at the University of Toronto, primarily with Megan Frederickson, but also with Chelsea Rochman, David Sinton and unofficially, Stephen Wright, in everything to do with duckweeds and duckweed microbiomes (from mutualisms, to ecotoxicology, to engineering better tools, and experimental “evolution” and epigenetics).
“Salt marsh microbiomes are impacted by waterway restrictions in coastal Maine.” Heather Richard, M.S., PhD. Candidate in Ecology and Environmental Sciences, UMaine.
Heather joined the University of Maine in 2021 as a PhD student with the Maine eDNA program and studies the impacts of bridges and roads on microbial communities in salt marsh habitats. Her background in Ecology led her to pursue a career in informal environmental education for several years before getting a Master’s degree in Marine Biology from San Francisco State University studying biofilms on microplastics pollution. Upon returning to Maine in 2016 she led local research for a coastal non-profit organization and has since been dedicated to studying coastal environmental issues relevant to Maine. She has found a true passion in bioinformatic analysis and is eager to learn new tools for data analysis of all kinds.
“Genomic Strategies for Discovering Microbial Dark Matter using Metagenomics“ Scott Tighe, M.S., Senior Research Associate, Environmental Microbiome Engineering Research Group, Dept of Civil Engineering, University of Vermont. Scott Tighe is currently Senior Research Associate in the Environmental Microbiome Engineering Research Group in the Dept of Civil Engineering at the University of Vermont and past technical director of the UVM Genomics Facility. He has expertise in all areas of genomics, microbiology, and mycology with a specific focus on advancing the methods used for microbiome and metagenomic analysis. He specializes performing metagenomic analysis in global extreme sites such Greenland, Antarctica, Romania, Saudi Arabia, Crete, Tahiti, and the International Space Station to name a few and is currently the science lead of the Extreme Microbiome Project (XMP). Short Abstract: The ability to perform advanced genomic techniques on samples collected from extreme environments demands high performance reagents and techniques not commonly used in most labs today. For the past15 years, our projects have developed novel sampling and DNA sequencing protocols for profiling a variety of environments, including snow, glaciers, Antarctic lakes, Romanian caves, thermophilic ecosystems, halophilic, acidic, alkaline lakes, anaerobic digestors as well as organisms from bioaerosols and the International Space Station. Technical approaches include novel liquid concentrating devices, sample disruptors, biopulverizers, and field use of the Oxford Nanopore DNA sequencer. Whole Genome sequencing results indicate that for many unique sights, 5-30% of all organisms are not classified beyond Genus with a large number being new species.
“A Sample-to-results Workflow for Sequencing from Wastewater“, Dr. Siva Chavadi, PhD. Senior FAS, NEBNext Products, NEB
Speaker Bio: Siva Chavadi (he/him) is a Senior FAS (NEBNext NGS sample prep reagents) at New England Biolabs, working with academic and core facilities as well as industrial collaborators to implement scalable NGS workflows. Previously at GENEWIZ/Azenta, he supported NGS service operations and customer applications. He holds a PhD in Animal Sciences from University of Hyderabad, India with research focused on TB diagnostics and vaccine development using functional genomics tools.
2:45 – 3:00 Break
3:00 – 4:00 Session 3: Practical Applications
“Antibiotic-Free Liquid Surface Coatings for the Tunable Control of Protein and Microorganism Adhesion: The rise of antibiotic resistance is one of the greatest global public health challenges of our time”, Dr. Caitlin Howell, PhD, Associate Professor of Bioengineering, Department of Chemical and Biomedical Engineering, University of Maine
Caitlin Howell is an Associate Professor of Bioengineering in the Department of Chemical and Biomedical Engineering at the University of Maine. She received her MS in Biology from UMaine, her PhD in Physical Chemistry from the University of Heidelberg in Germany, then completed a postdoc at the Wyss Institute for Bioinspired Engineering at Harvard University before returning to UMaine to found the Biointerface Engineering Lab in 2016. Her research group is focused on designing surfaces to leverage the power of living systems to address human problems.
“From Lab to Tap: Quality Control in the Craft Beverage Industry”, Sam White, M.S., Quality Control Collaboratory (QC2), University of Southern Maine
Samantha White is a dedicated researcher and educator with extensive experience in analytical chemistry and the craft beverage industry. Sam manages the Quality Control Collaboratory at USM where she leads lab operations, quality control initiatives, research projects, and industry outreach programs.
“From Bugs to Breakthroughs: Sequence the Invisible Majority with QIAseq.”, Dr. Kevin W. Joseph, PhD, Genomics Field Application Scientist
Kevin graduated from University of Maine in 2012 with a BS in Microbiology and later acquired his PhD in Infectious Disease and Microbiology from the University of Pittsburgh. During his graduate work, he focused on HIV-1 single genome sequencing and integration site analysis of clonally-expanded, replication competent, HIV-1 proviruses in patients with long-term viral suppression. He developed the Individual Proviral Sequencing Assay (IPSA) to study these rare targets, which enables sequencing of full-length HIV-1 single-genome proviruses and their integration sites with high sequencing efficiency. Kevin has brought his knowledge and expertise to QIAGEN where he assists researchers with NGS, PCR assay and oligonucleotide design, and in-lab training/support.
“Illumina innovation roadmap“, Jennifer Mosher, Senior Sequencing Specialist, Illumina
Jennifer Mosher is a Senior Sequencing Specialist at Illumina, with deep expertise spanning field applications, marketing, and NGS-based proteomics. With over a decade in academia, she previously led operations for NGS at the Cornell Genomics Core before transitioning to industry, where she partners with both academic and commercial leaders to drive adoption of advanced sequencing technologies and assays.
4:00 – 5:00 Posters and networking
Agenda for June 12, 2026
This workshop broadly includes combining microbiome data with other data types, combining qualitative and quantitative data, learning to evaluate microbiome data using demographic data, dealing with unbalanced groups especially in human microbiomes, and how to choose statistical tests for microbiome projects.
8:00 – 9:00 Breakfast, provided
9:00 – 10:00 Case Study Presentations, hosted by Sue Ishaq
“Human data pitfalls” by Kevin Roberge, M.S.. UMaine: Lecturer, Mathematics and Adjunct Professor, Women’s, Gender & Sexuality Studies.
“Statstical decisions” by Laura Jackson, Ph.D., UMaine: Bioinformatics Program Coordinator & Graduate FacultyGraduate School of Biomedical Science & Engineering, Bioinformatician – CORE Strategic Operational Services.
“A case study in human microbiomes”, by Lola Holcomb, Ph.D., University of New England, Bioinformatics Postdoc
10:00 – 10:15 Break, snacks and coffee provided
10:15 – 11:00 Panel discussion, symposium speakers are welcome to join
11:00 – 12:00 Open working time for data wrangling advice and analysis workflow storyboarding as a group
Kevin Roberge, can help with analysis storyboarding, identifying assumptions, math
Laura Jackson, can help with coding, statistics and deciding which to use, RNAseq data, 16S data, meta-analyses
Lola Holcomb, can help with coding, metagenomics data, 16S data, meta-analyses
Sue Ishaq, can help with some 16S, but will be helping to coordinate the workshop
12:00 – 1:30 lunch, provided
We are grateful to our financial supporters, who helped make this symposium possible and accessible!
Future Microbiome Researcher Sponsors
These incredible sponsors not only helped support this symposium, but their generous donation helped to create several hands-on laboratory workshops to support training in microbiome technology!
Congratulations to Ashley Reynolds and Ethan Glenn for their first publication!! This extensive literature review was the result of more than a year of dedicated researching, writing, and revising to synthesize existing knowledge on how diet can affect neuroinflammation and how gut microbes are involved. Titled “Plant-Derived Bioactives, the Gut–Brain Axis, and Neurodegenerative Diseases: Mechanistic Roles of Diet–Microbiota Interactions“, the review is available open-access here in the journal Frontiers in Neuroscience!
Lab work on diet and neuroinflammation
Ashley and Ethan’s review was a precursor to some of the lab work they’ve been doing in my and Yanyan’s lab, respectively, as well as in collaboration with Brigitte Lavoie at UVM, with whom we have worked for several years on mouse models of inflammation.
Ashley has an extensive background in nutrition and dietetics (bio is below), and joined our labs to understand how dietary metabolites and gut microbiome metabolites could be used to improve health, and reduce inflammation in the gut and the brain (neuroinflammation). She is a Doctor of Philosophy candidate in Human Nutrition and Food Sciences, and previously completed her undergraduate degree in Food Science and Human Nutrition in 2021 as a Maine Top Scholar, her Master’s degree in Food Science and Human Nutrition with Jade McNamara, at UMaine on intuitive eating in college students, and completed a dietetic internship and shortly after passed her RD exam to become a registered dietitian in 2023. She is incredibly interested in nutrition therapy and is beginning her research looking into the microbiome and metabolomic pathways in the context of IBD.
Ethan Glenn is a master’s student at SUNY Binghamton University School of Pharmacy in Yanyan Li’s Lab. Ethan has been helping to develop several models of neuroinflammation to use for testing the efficacy of different preparations of broccoli sprouts in delivering bioactives.
Ashley and Ethan have been collaborating on protocol design and testing, sample processing, and data analysis for several projects, including one we recently completed which will form the basis of Ashley’s dissertation.
Johanna, Ashley, Alexis, and Sue put in long hours during the mouse trial to collect samples each day.
1 School of Food and Agriculture, University of Maine, Orono, Maine, USA 04469
2 School of Pharmacy and Pharmaceutical Sciences, SUNY Binghamton University, Binghamton, New York, USA 13902
3 Dept. of Neurological Sciences, University of Vermont College of Medicine, Burlington, Vermont, USA 05405
Abstract
Diet is increasingly recognized as a potential upstream modulator of the gut-brain axis (GBA) through its effects on the microbiome, microbial metabolites, and host immune and endocrine responses. The GBA is a complex, bidirectional network connecting the gastrointestinal tract and central nervous system, with diet influencing microbial community structure and metabolic output. Plant-based diets, such as Mediterranean and MIND, have been associated with increased production of anti-inflammatory microbial metabolites and improved barrier function, while high calorie/low nutrient diets are often linked to increased immune activation and barrier dysfunction. However, while microbial metabolites, especially short-chain fatty acids, indoles, bile acids, and isothiocyanates, have been proposed as mediators of neuroprotective effects, their role in neurodegenerative diseases remains an area of active investigation, with evidence largely derived from preclinical and associative human studies. Cruciferous vegetables, especially broccoli sprouts, are an emerging focus of research for their bioactive compound sulforaphane, which activates Nrf2-centered cytoprotective pathways. Animal and early human studies suggest sulforaphane can improve cognitive and behavioral outcomes, though larger clinical trials are needed. Personalized, microbiota-targeted dietary interventions may offer scalable strategies for managing neuroinflammatory and neurodegenerative conditions, and we emphasize the need for integrated research across diet, microbiome, and brain health.
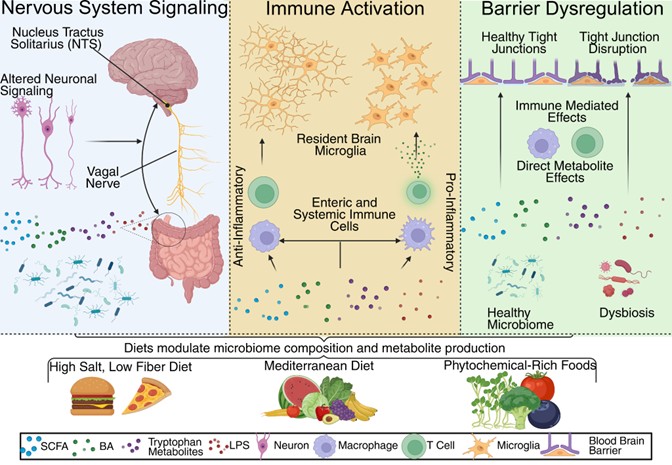
Figure 1: Mechanisms of how Diet Influences the Gut Brain Axis. Depicting three mechanisms of the gut brain axis of importance for dietary interventions. Different types of diets can modulate the microbiome to produce different metabolites. These metabolites can affect nervous system signaling through the ENS and vagal nerve. Immune activation via these metabolites either enterically or systemically can lead to activation of resident brain microglia. Dietary metabolites have also been shown to either protect blood brain barrier tight junctions or disrupt them, both through direct effects and immune mediated effects. Made in Biorender under license by E. Glenn.
We’ve all felt the thrill of synchronicity when meeting someone for the first time and realizing how we have much in common, but when this occurs for a dozen people simultaneously, who go on to share ideas and excitement for 13 hours straight, it’s magic. Thus, Professor of Anthropology, Roberta Raffeta’, created magic when she invited a group of microbiome and health science researchers together for the “Integrating Qualitative and Quantitative Data in Microbiome Research and Postgenomics: Toward an Interdisciplinary Dialogue” workshop at the Università Ca’ Foscari Venezia. Roberta works with many disciplines, including microbiome, computational, health, and social sciences, and her work often focuses on how research is designed, implemented, and interpreted. Her work across disciplines gives a larger view on how different disciplines approach similar research, as well as provides her with a rich network of colleagues.
The workshop was in the spirit of a project that Roberta, Prof. Nicola Segata, Prof. Elena Bougleux, and others investigating the sharing of microbes at Antarctic stations based on social interactions and shared spaces. That project is a clear example of how social systems can determine who you interact with, where, and how, and thus which microbes might get shared between you or between everyone at the base. While those data were still being processed and not shared, we did get to hear a bit about the journey to Antarctica from Elena.
After introducing the project, workshop, and ourselves, we began with a presentation by Professor Federico Russo. Her talk focused on how health is quantified, and how that alters the way we design and perform health research. Health is a complex concept that has biological, chemical, physical, and social aspects which can be measured and metric-ified. For example, there are bio-logical versus bio-social metrics of health (social effects of disease, feelings about health and outlook), and everything in between which can be measured to assess the state of health or disease. In addition, health research uses the term “social determinants”, which is similar to what biologists, microbiologists, or ecologists mean when using the term “environmental factors”, but these often refer to the same thing. These factors are the collection of host, social/community, environmental, and geological features which affect who you are and what you encounter in your life. Some of them seem very specific, like age, but the expression of age can be modified by staying active, eating well, managing stress, and avoiding pollution, so on its own, knowing someone’s age might not be useful information. Thus, to study health, we use both markers, like age, to tell us potential outcomes or indicators of one’s health state, and social determinants, like lifestyle features, to tell us possible causes or mitigating factors of health.
Yet, one cannot slice the concept of health into a thousand measurable factors and then expect to re-assemble them back into the concept it was – because what makes us feel well or healthy is not necessarily having or knowing that our biological metrics are good, it’s feeling well even if our biological markers are out “normal” parameters or when, on paper, we are sick. This brings up a concept that I had discussions about within this workshop group, with the faculty at Ghent, and with researchers through MSE and MiSt: that your social factors and the support network around you strongly influence whether you feel well or unwell, regardless of what your biological markers would suggest.
There is also a focus in healthcare on regaining health when someone is sick, with social or institutional support system for that (rehabilitation clinics, etc.), but there is not always an institutional focus on understanding how people stay healthy, in part because this is seen as a personal choice and not a result of adequate access to public resources (fresh food, water, air, shelter, education, safety), or as a function of useful public health policies which make is easy for people to take care of themselves. Simple features like sidewalks, bike paths, local grocery stores, free public restrooms, shade and places to sit, are all features that allow people to stay active, get around, stay healthy, and use their public spaces.
My research talk was next, and I focused on the steamed broccoli sprout intervention trial I completed a few years ago with Yanyan Li. That was a pilot study, which recruited 20 people to steam and eat broccoli sprouts every day for a month, to measure any changes in the gut microbiome, the metabolites it was producing, and whether gut bacteria would convert the inactive glucoraphanin in sprouts into the anti-inflammatory compound sulforaphane. Rather than focus on the microbiological, metabolomic, or diet survey results, I presented everything which went unexpectedly in the study, and what people told us about the challenges to consuming daily sprouts. This, in fact, was the real goal of the study, to understand which aspects of the diet would be challenging, or rewarding, and to try and make things as easy as possible. My observations on the diet study sparked excellent discussion, which gave me plenty of ideas on framing the scientific manuscript and what we learned from our participant’s data, as well as a perspective piece on the design and implementation of the study and what we learned from our participants’ feedback (not their data).
Another study observation which was also a reoccurring workshop discussion was the need for health studies that start with people’s perspectives and patient’s identification of problems, which then work backwards to understand how the microbiome is involved. This style of research is case-study and health engineering research to test applied research questions, and is needed in addition to the large-scale, double-blind experiments to test basic research questions. We talked at length about how most large-scale diet surveys are inadequate, no matter how detailed they are, because they become so vague as to be useless when they are generalized to ask about all possible food item diets. Most diet surveys that are meant to be broad ask for too much detail about things which are considered superfluous for individual research projects, and too little detail on critical info. For example, most diet surveys that gather diet history (eating habits over the last 6 – 12 months) are underpowered to assess fermentation products, don’t ask how people cook and make decisions about diet, and are quantified to assess compliance to an idealized and single idea of a healthy diet, even if it doesn’t work for every person (ex. dairy is good on many diet surveys but there is no place to select that you don’t consume dairy because you are allergic to it).
This discussion carried across lunch, during which we diverged into many animated conversations only to bring it back to quality of information in the presentation after lunch, by Professor Lisa Lehner, a health researcher who presented reflections on three research projects, in which the availability and completeness of data about patients was lacking, and this stymied researchers’ attempts to understand public health for three disease models, such as how these diseases are transmitted, how people access health care, and how migration, homelessness, or simply traveling often can impact access to care. For example, in trying to study human papilloma virus cases, her research found that country of sampling was included in patient records but not country of infection. Similarly, the idea of “where do you live”, “where are you from”, or “where have you been recently where you might have contracted this infection” are very different questions with different contexts to the answers, not all of which will provide useful information for this specific study depending on which the patient was asked and what their history was.
Information sharing is both the key and the challenge, and we discussed how often the information to resolve public health crises exists, but it’s not all in one place, it’s missing information, it doesn’t contain the right context, institutions don’t want to share, some information is private or access is limited even to medical providers, some information is not retained, and even when we get much of this in one place the amount of detail is overwhelming to the point where researchers need to spend years trying to figure out how to make it useful – what’s important to know? What is a ‘red herring’? — before it even gets to be used for infection tracking, treatment, and prevention. Her research also highlighted inequities in healthcare, and that sometimes information that would help us understand infection data can’t be made public, because you might reveal information about sensitive populations that can be used for discrimination. For example, people without health insurance might be migrants that are no longer on active visas, and quantifying how many people can make them a political target.
The discussion after the talk was so engaging, and blended into the focus of another speaker, so we informally heard from Professor Donato Giovannelli, who was not able to present his talk (yet!) because the workshop was running later than expected (we all had so much to talk about!) and he had to catch a train. Donato is a geomicrobiologist examining microbes in extreme environments to understand how they survive and function. He has a particular focus on the microbial fixing of gaseous hydrogen which produces water, and how biological sinks for freshwater (like us walking, talking, water bottles), actually allow for the preservation of large quantities of water on Earth. Donato’s short version of his talk was focused on the scale of standardization in research. He argued that what works in the lab or the field changes based on the needs and circumstances of each project, so you need to thoroughly describe what you did but that doesn’t mean every lab uses, or has to use, the exact same protocols, kits, or methods every time. In fact, even if it were possible to replicate circumstances exactly, trying to do everything identically will just make all studies biased in the same ways. There is no way to replicate some experiments, especially with humans, because no human is ever replicable, even to ourselves.
Highlighting the importance of methods and process in research brought up another challenge in research: it is important to include vocabulary to describe what you mean, in addition to what you did, but Methods sections are often compressed by scientific journal word limits, and all the nuance and context or the problem-solving portion, gets cut for space and is over compressed to the point of being useless. Donato argued, as many scientists do, that the explanation of the methods should be the majority of the paper, and many labs do publish the long form of the methods as a co-published paper, or as a protocol paper that gets cited in the whole scientific experiment paper. Our workshop heartily agreed. Science is a process, as scientists we are creating this process and this is our most valuable contribution, more so, even, than the results.
Sometimes the drive for standardization also leaves out people who contributed to the study in a way, but their contribution is considered “not science”. For example, technicians, field personnel, or anyone who helped you access or collect samples but did not work on the samples or contribute “conceptual framing or interpretation” don’t count as authors under most journal guidelines, even if you could not have done the project without them. Similarly, if you designed your project based on feedback and direction from the public or specific audiences, this is somehow not considered “conceptualization” or experimental design (???!!!). It is always up to the author team to decide who is and isn’t an author, but without more flexible guidelines on authorship, it can be easy to remove someone’s contribution to a publication.
This type of value judgement in science segued us to our last presentation of the day, on how the terms/definitions we use, the factors we choose to study, the things we prioritize in our research, all reflect values we have placed on some things over other. It can also, unintentionally and intentionally, place inherent value on having certain study results or outcomes. And when the study is about people, placing value judgements on biological, microbiological, or social factors can create a hierarchy – you can see where I am going with this – in which certain people are implied to have superior metrics. This is something a research team and I published on in microbiome research, and has been the focus of a multi-year project led by Professor Abigail Nieves Delgado, Dr. Aline Potiron, and others, on how microbiome scientists define terms which are used to describe people, and how those definitions carry assumptions that may influence the interpretation of the results by researchers or readers.
Population descriptors are ways of, well, describing populations. However, the selection of terms, how you define them, how you categorize those terms, are often too strict to be applied to real-life people or in different communities. Using the same example of age, how do you compare age in two populations with wildly different exercise, diet, or stress load which all impact the physical or behavioral expression of age? More to the point of this talk, how do you categorize ancestry without nuance? Do you focus on genomics, or culture, or both? Do you place people in categories based on certain factors, or let them choose their own? How do you compare people in one location to another without considering the history of war, colonization, and slavery had on the ability of people to choose with whom to have children? And does ancestry even matter when you consider the myriad changes to health and the microbiome effected by what you eat, what animals and people you interact with, and where you live? Scientists all working on the human microbiome had wildly different understanding or definitions of population descriptors, and beliefs around what population descriptors were caused by or caused changes to the microbiome. And this, despite most microbiome studies having little to no need for information on ancestry or race to answer their research question.
Studies don’t gather data on every possible factor in people, that would require a detailed biography of every participant. Researchers choose what type of data to collect about someone based on what is important to the study and answering the research question – but as researchers we tend to think within the constraints of our own discipline – what info do I need for my statistics or to publish in my field? When that happens, we compress the variability of the people we study into fewer dimensions, example like taking an in-person experience at the beach and representing it as the words “seashells”, “gritty floor”, and “wet”.
Finally, a theme that came up several times during the workshop was that human social structure or institutions sometimes have so many constraints as to fail in their mission to provide resources to the public. As a perfect example, we did not have air conditioning at the university, even though was 85 degrees outside and between 90 and 105 degrees F inside some of the rooms after we had been in them for a while, because it is university policy to turn air conditioning on for the whole campus only after a certain date regardless of what is going on in real life. I don’t want to pick on the university, because this is a very common policy at universities, including UMaine. But it highlights the point that solutions (to air conditioning, microbiomes, health, etc.) need to be nimble enough to respond to local conditions using the input of people who are actually living through those conditions. And in this way, we have the crux of the workshop: how do we perform local-conscious and nimble science that is driven by the needs of actual living things or ecosystems, against the tide of institutional habit? For me, that’s going to involve more community-based and person-driven research questions, more collaboration with this remarkable group of researchers, and hopefully a lot more fresh, Italian mozzarella.
With a suitcase full of chocolate and a notebook full of ideas, I said goodbye to Ghent, Belgium after a 4-day visit. I was generously invited here to give a seminar to the Interdisciplinary Medical and Health Seminar series which is co-organized by the Department of Public Health and Primary Care and the Department of Internal Medicine and Pediatrics, and to brainstorm about nutrition and food science, health equity, and gut microbiomes.
Ghent University is health sciences focused, and their departments range from public health, internal medicine, surgery, oncology, and every practical application of medicine one could think of. They have ethics and equity departments, and have some of the leading programs that specialize in care and policies for trans health as well as for refugee populations. Many departments included research on inflammation and Inflammatory Bowel Diseases, as well as digestion and gut microbiomes, as so many aspects of health and disease are connected to gut health. But, the integration of food science and using nutritional intervention was somewhat sparse – thus my invitation to give a talk, as the UMaine School of Food and Agriculture (which I am part of) programs in Nutrition, Dietetics, and Food Science (which I am affiliated with), as well as my collaborative work with Dr. Yanyan Li on broccoli sprouts and the gut microbiome, all emphasize the integration of diet into health plans.
The seminar was well attended, and I appreciated how many students were able to join despite being so close to final exams. I always appreciate sharing the work that I’ve been doing with collaborators on broccoli sprout bioactive and the gut, as well as with Microbes and Social Equity, and more recently, the Microbiome Stewardship and IUCN Microbe Specialist groups on conservation and policy. The real treat, though, was the long conversations I had with faculty and students. I spent several days with Professor Sara Willems, and her PhD student Jarne Ghijsels, who are researching access to diet, nature, and healthcare in several populations including the Sami people who are often barred from performing their traditional migration, food systems, health practices, or diet. Sara’s work highlights the critical importance of holistic lifestyle changes and the involvement of family and community in achieving health. Jarne’s project examines how exposure to nature can be used to improve mental and physical health in patients as well as care givers, and how to integrate “nature prescriptions” into general medicine practice. Hopefully, Sara and Jarne will generously share their work with the Microbes and Social Equity speaker series next year!
In addition, I spent hours in engaging conversation with Professor Debby Laukens, whose expertise on neuro- and gut inflammation, as well as mouse models of colitis, was extremely informative for me, and who was very interested in the use of diet to reduce inflammation. I enjoyed the camaraderie of dinner with my hosts, PhD student Sarah Derveeuw, Prof Sorana Toma, and Professor Luisa Borrel (City University of New York), another guest speaker and expert of epidemiology and biostatistics.
Finally, I also wanted to acknowledge the gentle schedule that was set up for me. Overnight travel is never easy, and leaving Maine always requires driving to an airport and an extra flight to a larger hub. My hosts provided an entire day in which I could adjust, and Jarne picked me up from the train station to help me navigate the tram in my jet-lagged state. When I was going to have to wait most of the day to be able to check into my hotel, they made arrangements for me to shower on campus, and when my flight arrived much later than scheduled, Jarne pivoted to help me check in, get lunch, and plan my travel to campus. My meetings were scheduled mid-morning instead of at 7 or 8 AM, and they set aside plenty of breaks. The relaxed schedule was a perfect start to my week-long trip, and allowed me time to relax, think creatively about science, and, as always, to keep working on my primary responsibilities which never take a day off (last week we had unexpected student bills, posters to prepare for conferences, mouse diet to make, a mouse trial to finish coordinating, a proposal to write, and … of yes, blog posts to write).
In between meetings, I, of course, took advantage of my free time to walk around Ghent, which is one of the cities with the best-preserved Medieval architecture, but is also forward-thinking in its design and supports pedestrian, bike, and public transport over personal cars. The effect was stunning: downtown there was very little air pollution from traffic, I could safety walk everywhere, and by going on foot I was able to appreciate the little details, such as the wrought iron, murals, or frescos on many of the buildings. There were plenty of little parks and places to sit in the shade and appreciate the breeze off the canals. Walking also helped counteract all the chocolate and cheese, which are two of the primary attractions in Belgium 🙂
After leaving Ghent, I made my way to Italy, where I am presenting my work again, this time with a focus on combining human subjects data, survey data, and microbiome data from the pilot project I completed a few years ago with Yanyan. Ciao, Venezia!
Last week, we completed our third sequencing workshop with Illumina, and we are grateful to Katherine Sullivan, Cameron Robertson, Curtis Provencher, and Michelle Benitez for their patience, expertise, and encouragement. Their sponsorship provided the opportunity for 7 students, plus me, and Alex and Megan at UMaine CORE, to learn how to prepare their samples for sequencing.
We are also grateful to Alex Sacco, Genomics Facility Manager at UMaine, as well as UMaine CORE, for helping to organize the workshop and ensure that students had access to specialized equipment and supplies. Alex and I have been co-organizing a series of workshops, and we hope to make these a regular part of genomics education at UMaine.
We saved the most challenging type of sequencing for last: RNA sequencing. Unlike the double-stranded and protein-secured format of most DNA, RNA is single stranded which makes it susceptible to breaking or damage from high temperatures, freezing and thawing too quickly, acid or base, and the many enzymes that cells make to break RNA back down. RNA is the short-term copy of DNA that cells make as a carbon copy to take over to ribosomes to have them translate the RNA code and make a protein. Cells use RNA when they need to get things made, because proteins are how the cell accomplishes most of its functions, and when there is a demand they will make many RNA copies of the same gene to get many copies made into proteins. But once the cell needs something else, it recycles the RNA, and it is because RNA is a disposable tool that it is so easily damaged can be extremely tricky to obtain. RNA is also a very valuable source of information about how a cell is acting or reacting, so it’s worth the trouble. As such, the lab work needed to extract RNA from cells has more steps and requires more delicacy than the process to extract DNA from a cell.
Illumina sequencing platforms can be used for RNA or DNA, and they sell kits that contain the supplies and reagents needed to target specific types of RNA, such as the ones that animals make versus the ones that microbes make. In this way, researchers can figure out how animal or human cells act and how their microbial communities are acting. Illumina helped us learn how to isolate host RNA, using intestinal tissues from mice from a recent neuroinflammation and broccoli sprouts trial, as well as tissues from several organs on a squid!
Graduate and undergraduate students in agriculture, microbiology, and ecology gathered to learn the process of extracting, cleaning, preserving, sequencing, and of course, troubleshooting, over a two-and-a-half day workshop led by experts Katherine, Cameron, Curtis, and Michelle. Their infinite patience was greatly appreciated, as our students and I were all very new to working with RNA and we all had a lot to learn. We can’t wait to see what the data looks like, and to use it to fine-tune our workflow for going back to process the rest of the intestine tissues to make sure we succeed in getting the RNA we are looking for.
Most of the students in the workshop are pictured here, along with Illumina and UMaine CORE sequencing experts.
Whether we are interested in the way coastal wetlands sequester carbon, how diet affects us differently, or how public and environmental health are inextricably linked – research on the microbiome can reveal how systems connect. Join us for two days to learn about how microbial communities impact ecosystems, food production, health, and more; to hear from experts researching these issues in New England and beyond; and discuss the technology and data analysis which can boost your own research.
UMaine Buchanan Alumni House, 160 College Ave, Orono, ME 04473
If you would like to help sponsor this event, please contact sue.ishaq@maine.edu.
Agenda for June 11, 2026
8:00 – 9:00 am Breakfast, provided
9:00 – 9:15 Welcome and Opening Remarks
Dr. Sue Ishaq, PhD, Associate Professor of Microbiomes, Associate Director of Special Projects in the School of Food and Agriculture at the UMaine; member of the Board of Directors at ASM; Founder and Lead of the Microbes and Social Equity working group.
9:15 – 10:15 Plenary Presentation
“The second messenger c-di-GMP in Bordetella”
Dr. Federico Sisti, Ph.D., Investigator, Institute of Biotechnology and Molecular Biology (IBBM) of the National Scientific and Technical Research Council (CONICET). Dr. Sisti is incoming President-Elect at the American Society of Microbiology, and the first ASM President representing its global membership.
10:15 – 10:30 Break
10:30 – 12:00 Session 1: Microbial mayhem or tiny partners in health?
“Leveraging organ-on-chip technology to study gut microbiome effects on human health and disease“, Dr. Dani Brassino, PhD, Assistant Professor of Gut Microbiome and Cancer Interactions, The University of Vermont College of Medicine.
Following a B.S. in Nutrition Science and at the University of Texas, Dani Brasino acquired a PhD in Chemical Engineering at the University of Colorado. At CU, she developed synthetic phospholipids as part of an artificial cell project and acquired experience in microfluidics fabrication. Combining these experiences, she led development of a novel polycarbonate-based organ-on-chip platform as a postdoc at Oregon Health and Science University. Now her lab, the μMicrobiome Lab, aims to further develop and apply gut microbiome-on-chips to study the relationship between the human gut microbiome, distal disease progression, and therapeutic efficacy.
“Genetic and Microbiome Control of Addiction-like Behaviors in Mice”, Dr. Jason Bubier, Ph.D., Senior Research Scientist, Center for Addiction Biology, The Jackson Laboratory
Jason A. Bubier, Ph.D., is a Senior Research Scientist at The Jackson Laboratory, where he leads research at the intersection of genetics, behavior, and addiction biology. His work focuses on uncovering the genetic and physiological factors that shape vulnerability to addictive substances, with current projects examining the microbiome’s influence on behavior, the genetic architecture of opioid‑induced respiratory depression, and systems‑level pathways involved in addiction.
Dr. Bubier has spent more than two decades at The Jackson Laboratory, advancing through multiple research roles and contributing to major initiatives in functional genomics and behavioral genetics. He has authored more than 80 scientific publications, and his work is widely cited across fields including immunology, bioinformatics, and addiction science.
“Food insecurity, gut microbiome, health”, Dr. Maria Carlota Dao, Ph.D., Assistant Professor of Human Nutrition, University of New Hampshire.
Dr. Maria Carlota Dao is an Assistant Professor of Human Nutrition in the Department of Agriculture, Nutrition, and Food Systems at the University of New Hampshire. As an interdisciplinary scientist focused on obesity research, she investigates the interplay of cardiometabolic risks, dietary and psychosocial factors, and the gut microbiota. Through this work she seeks to address obesity in health disparity populations. Prior to becoming a faculty member at UNH, Dr. Dao worked as a scientist at the Jean Mayer USDA Human Nutrition Research Center on Aging at Tufts University. Her training includes a postdoctoral fellowship at the Sorbonne University and INSERM (Institute of Cardiometabolism and Nutrition) in France, and a PhD in Biochemical and Molecular Nutrition from the Friedman School of Nutrition Science and Policy at Tufts University.
“Gut Anthro“, Dr. Amber Benezra, Assistant Professor of Science and Technology Studies at Stevens Institute of Technology
Dr. Benezra is a sociocultural anthropologist researching how studies of the human microbiome intersect with biomedical ethics, public health/technological infrastructures, and care. In partnership with human microbial ecologists, she has been developing an “anthropology of microbes” to address global health problems across disciplines. Her book, Gut Anthro (published by University of Minnesota Press in 2023) is the first ethnography of the microbiome.
“One Codex in Action“, Christopher Smith, One Codex.
12:00 – 1:15 Lunch, provided
1:15 – 2:45 Session 2: Biochemical geniuses: microbes from soil to sea
“How to build a microbiome to “breed” better plant-inoculants.” Dr. Anna O’Brien, PhD., Assistant Professor of Molecular, Cellular, and Biomedical Sciences, University of New Hampshire.
She is originally from the Pacific Northwest. She received her B.S. in Plant Biology from the University of Washington, her PhD from the University of California Davis, where she worked with Jeffrey Ross-Ibarra and Sharon Strauss on the impacts of rhizosphere biota on trait divergence and adaptation in the wild relatives of maize. Anna Post-doc’ed at the University of Toronto, primarily with Megan Frederickson, but also with Chelsea Rochman, David Sinton and unofficially, Stephen Wright, in everything to do with duckweeds and duckweed microbiomes (from mutualisms, to ecotoxicology, to engineering better tools, and experimental “evolution” and epigenetics).
“Salt marsh microbiomes are impacted by waterway restrictions in coastal Maine.” Heather Richard, M.S., PhD. Candidate in Ecology and Environmental Sciences, UMaine.
Heather joined the University of Maine in 2021 as a PhD student with the Maine eDNA program and studies the impacts of bridges and roads on microbial communities in salt marsh habitats. Her background in Ecology led her to pursue a career in informal environmental education for several years before getting a Master’s degree in Marine Biology from San Francisco State University studying biofilms on microplastics pollution. Upon returning to Maine in 2016 she led local research for a coastal non-profit organization and has since been dedicated to studying coastal environmental issues relevant to Maine. She has found a true passion in bioinformatic analysis and is eager to learn new tools for data analysis of all kinds.
“Genomic Strategies for Discovering Microbial Dark Matter using Metagenomics“ Scott Tighe, M.S., Senior Research Associate, Environmental Microbiome Engineering Research Group, Dept of Civil Engineering, University of Vermont. Scott Tighe is currently Senior Research Associate in the Environmental Microbiome Engineering Research Group in the Dept of Civil Engineering at the University of Vermont and past technical director of the UVM Genomics Facility. He has expertise in all areas of genomics, microbiology, and mycology with a specific focus on advancing the methods used for microbiome and metagenomic analysis. He specializes performing metagenomic analysis in global extreme sites such Greenland, Antarctica, Romania, Saudi Arabia, Crete, Tahiti, and the International Space Station to name a few and is currently the science lead of the Extreme Microbiome Project (XMP). Short Abstract: The ability to perform advanced genomic techniques on samples collected from extreme environments demands high performance reagents and techniques not commonly used in most labs today. For the past15 years, our projects have developed novel sampling and DNA sequencing protocols for profiling a variety of environments, including snow, glaciers, Antarctic lakes, Romanian caves, thermophilic ecosystems, halophilic, acidic, alkaline lakes, anaerobic digestors as well as organisms from bioaerosols and the International Space Station. Technical approaches include novel liquid concentrating devices, sample disruptors, biopulverizers, and field use of the Oxford Nanopore DNA sequencer. Whole Genome sequencing results indicate that for many unique sights, 5-30% of all organisms are not classified beyond Genus with a large number being new species.
“A Sample-to-results Workflow for Sequencing from Wastewater“, Dr. Siva Chavadi, PhD. Senior FAS, NEBNext Products, NEB
Speaker Bio: Siva Chavadi (he/him) is a Senior FAS (NEBNext NGS sample prep reagents) at New England Biolabs, working with academic and core facilities as well as industrial collaborators to implement scalable NGS workflows. Previously at GENEWIZ/Azenta, he supported NGS service operations and customer applications. He holds a PhD in Animal Sciences from University of Hyderabad, India with research focused on TB diagnostics and vaccine development using functional genomics tools.
2:45 – 3:00 Break
3:00 – 4:00 Session 3: Practical Applications
“Antibiotic-Free Liquid Surface Coatings for the Tunable Control of Protein and Microorganism Adhesion: The rise of antibiotic resistance is one of the greatest global public health challenges of our time”, Dr. Caitlin Howell, PhD, Associate Professor of Bioengineering, Department of Chemical and Biomedical Engineering, University of Maine
Caitlin Howell is an Associate Professor of Bioengineering in the Department of Chemical and Biomedical Engineering at the University of Maine. She received her MS in Biology from UMaine, her PhD in Physical Chemistry from the University of Heidelberg in Germany, then completed a postdoc at the Wyss Institute for Bioinspired Engineering at Harvard University before returning to UMaine to found the Biointerface Engineering Lab in 2016. Her research group is focused on designing surfaces to leverage the power of living systems to address human problems.
“From Lab to Tap: Microbial Management in the Craft Beverage Industry”, Sam White, M.S., Quality Control Collaboratory (QC2), University of Southern Maine
Samantha White is a dedicated researcher and educator with extensive experience in analytical chemistry and the craft beverage industry. Sam manages the Quality Control Collaboratory at USM where she leads lab operations, quality control initiatives, research projects, and industry outreach programs.
“From Bugs to Breakthroughs: Sequence the Invisible Majority with QIAseq.”, Dr. Kevin W. Joseph, PhD, Genomics Field Application Scientist
Kevin graduated from University of Maine in 2012 with a BS in Microbiology and later acquired his PhD in Infectious Disease and Microbiology from the University of Pittsburgh. During his graduate work, he focused on HIV-1 single genome sequencing and integration site analysis of clonally-expanded, replication competent, HIV-1 proviruses in patients with long-term viral suppression. He developed the Individual Proviral Sequencing Assay (IPSA) to study these rare targets, which enables sequencing of full-length HIV-1 single-genome proviruses and their integration sites with high sequencing efficiency. Kevin has brought his knowledge and expertise to QIAGEN where he assists researchers with NGS, PCR assay and oligonucleotide design, and in-lab training/support.
“Illumina innovation roadmap“, Jennifer Mosher, Senior Sequencing Specialist, Illumina
Jennifer Mosher is a Senior Sequencing Specialist at Illumina, with deep expertise spanning field applications, marketing, and NGS-based proteomics. With over a decade in academia, she previously led operations for NGS at the Cornell Genomics Core before transitioning to industry, where she partners with both academic and commercial leaders to drive adoption of advanced sequencing technologies and assays.
4:00 – 5:00 pm Posters and networking
Agenda for June 12, 2026
This workshop broadly includes combining microbiome data with other data types, combining qualitative and quantitative data, learning to evaluate microbiome data using demographic data, dealing with unbalanced groups especially in human microbiomes, and how to choose statistical tests for microbiome projects.
8:00 – 9:00 am Breakfast, provided
9 – 10- ish am Case Study Presentations, 20 min each, hosted by Sue Ishaq
“Human data pitfalls” by Kevin Roberge, M.S.. UMaine: Lecturer, Mathematics and Adjunct Professor, Women’s, Gender & Sexuality Studies.
“Statstical decisions” by Laura Jackson, Ph.D., UMaine: Bioinformatics Program Coordinator & Graduate FacultyGraduate School of Biomedical Science & Engineering, Bioinformatician – CORE Strategic Operational Services.
“A case study in human microbiomes”, by Lola Holcomb, Ph.D., University of New England, Bioinformatics Postdoc
10 – 10:15 Break, snacks and coffee provided
10:15 – 11 Panel discussion, symposium speakers are welcome to join
11 – 12 Open working time for data wrangling advice and analysis workflow storyboarding as a group
Kevin Roberge, can help with analysis storyboarding, identifying assumptions, math
Laura Jackson, can help with coding, statistics and deciding which to use, RNAseq data, 16S data, meta-analyses
Lola Holcomb, can help with coding, metagenomics data, 16S data, meta-analyses
Sue Ishaq, can help with some 16S, but will be helping to coordinate the workshop
12 – 1:30 lunch, provided
We are grateful to our financial supporters, who helped make this symposium possible and accessible!
Future Microbiome Researcher Sponsors
These incredible sponsors not only helped support this symposium, but their generous donation helped to create several hands-on laboratory workshops to support training in microbiome technology!
As the academic year draws to a close (for the students), a flurry of awards ceremonies recognize their dedication and ingenuity. This year, as every year, the Ishaq Lab is grateful for all we have to celebrate! Only about a third of the lab was able to make it to our end of the semester good-bye party, but I look forward to their celebrating their continued successess in the lab and beyond.
Alexandra Ruff, Bella Murphy, Aaron Williams, Ashleys Reynolds, and Sue
Doctors
Johanna Holman, PhD., was recognized as an Outstanding Graduating Student for 2026 by the UMaine Graduate Student Government, and received her doctorate in Microbiology! This was actually her FOURTH award during her graduate studies. Johanna wasn’t able to have her doctoral hood bestowed at the UMaine graduate student graduation, as she just flew to Birmingham, England to start a postdoc position in the lab of Dr. Lindsay Hall, who focuses on early life microbial exposure and health.
Holman, Johanna. “The Metabolism And Community Contributions Of A Gut-Derived, Glucoraphanin-Degrading Bacteria: A Multi-Omics Approach.” (2026). University of Maine. Doctor of Philosophy in Microbiology. (Forthcoming).
Aaron Williams, B.S. Zoology, was awarded Honors for his thesis! Aaron started in the lab for a bit his first year, and came back in his last two years to complete his Honors research thesis on bacterial metabolism of glucoraphanin as a means of surviving acidic conditions, as well as effect on the byproducts produced. Here we are posing with the spectrophotometer that played a critical role in generating very large spreadsheets of data. Aaron is heading to Tufts Veterinary College in the fall!
Mia PoirierMiriam Talalay
Mia Poirier, B.S. Biology (Pre-Med Concentration) and Madison Ringuette, B.S. Animal and Veterinary Science (Pre-Vet Concentration), and Miri Talalay, B.S. Zoology and Veterinary Studies, are also graduating. Mia and Madison are both taking a year to gain medical and research experience before diving back into college for medical and veterinary programs, respectively. Over the last semester, Mia and Madison helped with two mouse trials investigating the effect of broccoli sprout diets on the gut microbiome, which can use the diet to make anti-inflammatories to reduce symptoms. The first was a pilot project testing our two of our bacterial cultures as probiotics, which is part of Alexis Kirkendall’s work, and the second was a project investigating the efficacy of broccoli sprout diets for resolving gut and neuroinflammation in young adult mice, which is part of Ashley Reynold’s work. Mis was awarded funding for her work in the lab: 2025/2026 Undergraduate Research Award from the UMaine Center for Undergraduate Research and the UMaine Institute of Medicine. Miri had been working us since her first year at UMaine! Miri worked on a variety of projects, helping provide key technical support on some of our years-long culturing projects. This included the screening of hundreds of bacterial cultures for their ability to metabolize glucoraphanin, and whether they would be able to use glucoraphanin and its byproducts to survive in acidic conditions (which Aaron also focused on).
All four Future Doctors will be featured as authors and contributors on our manuscript in development, led by Johanna, which will present the culturing work we’ve done over the last four years! We hope to get that submitted for peer review this summer.
Today was a blast at the Microbiome Workshop on Long-Read Amplicon Library Generation, the second in a series of hands-on workshops this spring that I have been coorganizing with Alex Sacco, Genomics Facility Manager at UMaine! This one was led by Alex, who is a genius at sorting out the complex math and coordination to combine multiple projcts with multiple targets onto one Oxford Nanopore Technology flow cell!!
Today we used materials generously provided by New England Biolabs, whos sponsorship provided the opportunity for 11 students to learn how to prepare their samples for sequencing. We are grateful to Siva Chavadi and Heidi Iuvino from NEB for their time, expertise, and support! NEB will be joining us on campus in June, too, for the Microbiome Symposium at UMaine.
We are also grateful to UMaine CORE, for helping to organize these workshops and ensure that students had access to specialized equipment and supplies.
During today’s workshop, students investigated the diversity of microbial communities by preparing sequencing libraries targeting bacterial, archaeal, and fungal marker genes. Using extracted microbial DNA which had been amplified with PCR, students learned how to ligate barcodes and combine samples into a pool for using the Oxford Nanopore MinION Nanopore Sequencing Student Workshop Protocol.
By amplifying and sequencing these variable regions, students generated data that can be used to compare microbial community composition across different sample types. Students who were at both workshops will also be able to compare the two sequencing strategies we have focusd on, which use different technology and provide unique perspectives during data analysis and interpretation.