
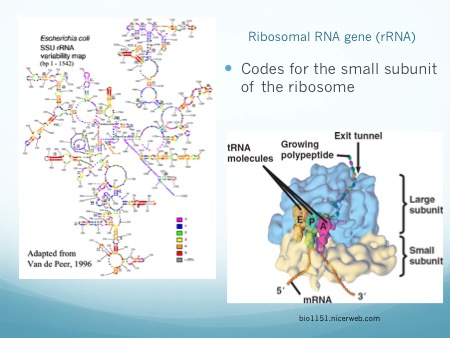
Microbiome studies do not usually employ culturing techniques, and many microorganisms are too recalcitrant to grow in the laboratory. Instead, presumptive identification is made using gene sequence comparisons to known species. The ribosome is an organelle found in all living cells (they are ubiquitous), and it is responsible for translating RNA into amino acid chains. The genes in DNA which encode the parts of the ribosome are great targets for identification-based sequencing. In particular, the small subunit of the ribosome (SSU rRNA) provides a good platform for current molecular methods, although the gene itself does not provide any information about the phenotypic functionality of the organism.

Prokaryotes, such as bacteria and archaea, have a 16S rRNA gene which is approximately 1,600 nucleotide base pairs in length. Eukaryotes, such as protozoa, fungi, plants, animals, etc., have an 18S rRNA gene which is up to 2,300 base pairs in length, depending on the kingdom. In both cases, the 16 or 18 refers to sedimentation rates, and the S stands for Svedberg Units, all-together it is a relative measure of weight and size. Thus, the 18S is larger than the 16S, and would sink faster in water. In both genes, there exist regions which are conserved (identical or near-identical) across taxa, and nine variable regions (V1-V9) [1]. The variable regions are generally found on the exterior of the ribosome, where they are more exposed and prone to higher evolutionary rates. Since the outside of the ribosome is not integral to maintaining its structure, the variable regions are not under functional constraint and may evolve without destroying the ribosome. They provide a means for identification and classification through analysis [2-6]. The conserved areas are targets for primers, as a single primer can bind universally (to all or nearly-all) to its target taxa. The conserved regions are all on the internal structure of the ribosome, and too much change in the sequence will cause its 3D (tertiary) structure to change, thus it won’t be able to interact with the many components in the cell. Mutations or changes in the conserved regions often causes a non-functional ribosome and will kill the cell.

In addition to a small subunit, ribosomes also possess a large subunit (LSU rRNA), the 23S rRNA in prokaryotes, and the 28S rRNA in eukaryotes. Eukaryotes have an additional 5.8S subunit which is non-coding, and all small and large units of RNA have associated proteins which aid in structure and function. Taken together, this gives a combined 70S ribosome in prokaryotes, and a combined 80S ribosome rRNA in eukaryotes.
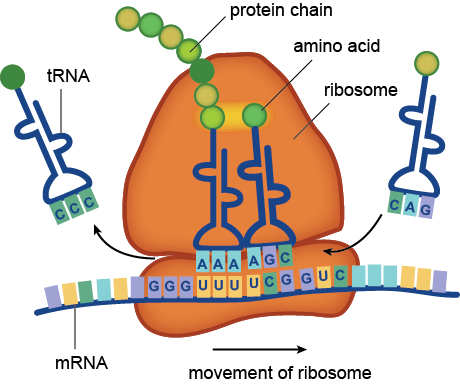
The way to study the rRNA gene is to sequence it. First, you need to extract the DNA from cells, and then you need to make millions of copies of the gene you want using Polymerase Chain Reaction (PCR). PCR and sequencing technology more or less work the same way as a cell would make copies of DNA for cell processes or division (mitosis). You take template DNA, building block nucleotides, and a polymerase enzyme which is responsible for reading the DNA sequence and making an identical copy, and with hours of troubleshooting get a billion copies! Many sequencing machines use nucleotides that have colored dyes attached, and when a nucleotide is added, that dye gets cut (cleaved) off, and the camera can catch and interpret that action. It then records each nucleotide being added to each separate DNA strand, and outputs the sequences for the microorganisms that were in your original sample!
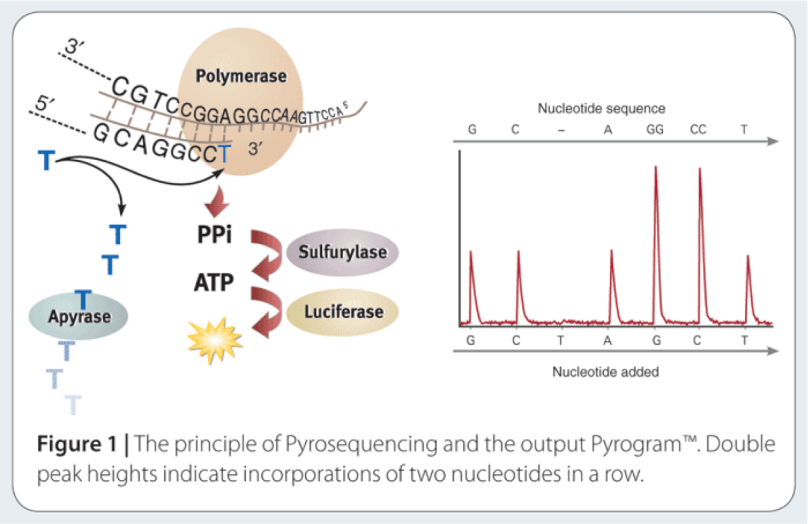
The two main challenges facing high-throughput sequencing are in choosing a target for amplification, and being able to integrate the generated data into an increased understanding of the microbiome of the environment being studied. High-throughput sequencing can currently sequence thousands to millions of reads which are up to 600-1000 bases in length, depending on the platform. This has forced studies to choose which variable regions of the rRNA gene to amplify and sequence, and has opened up an arena for debate on which variable region to choose [2]. And of course, the DNA analysis of all this data you’ve now created is quickly being recognized as the most difficult part- which is what I focused on during my post-doc in the Yeoman Lab. Stay tuned for a blog post on the wonderful world of bioinformatics!
- Neefs J-M, Van de Peer Y, Hendriks L, De Wachter R: Compilation of small ribosomal subunit RNA sequences. Nucleic Acids Res 1990, 18:2237–2318.
- Kim M, Morrison M, Yu Z: Evaluation of different partial 16S rRNA gene sequence regions for phylogenetic analysis of microbiomes. J Microbiol Methods 2010, 84:81–87.
- Doud MS, Light M, Gonzalez G, Narasimhan G, Mathee K: Combination of 16S rRNA variable regions provides a detailed analysis of bacterial community dynamics in the lungs of cystic fibrosis patients. Hum. Genomics 2010, 4:147–169.
- Yu Z, Morrison M: Comparisons of different hypervariable regions of rrs genes for use in fingerprinting of microbial communities by PCR-denaturing gradient gel electrophoresis. Appl Env Microbiol 2004, 70:4800–4806.
- Lane DJ, Pace B, Olsen GJ, Stahl DA, Sogin ML, Pace NR: Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci USA 1985, 82:6955–6959.
- Yu Z, García-González R, Schanbacher FL, Morrison M: Evaluations of different hypervariable regions of archaeal 16S rRNA genes in profiling of methanogens by archaea-specific PCR and denaturing gradient gel electrophoresis. Appl Env Microbiol 2007, 74:889–893.
