Congratulations to Ashley Reynolds and Ethan Glenn for their first publication!! This extensive literature review was the result of more than a year of dedicated researching, writing, and revising to synthesize existing knowledge on how diet can affect neuroinflammation and how gut microbes are involved. Titled “Plant-Derived Bioactives, the Gut–Brain Axis, and Neurodegenerative Diseases: Mechanistic Roles of Diet–Microbiota Interactions“, the review is available open-access here in the journal Frontiers in Neuroscience!
Lab work on diet and neuroinflammation
Ashley and Ethan’s review was a precursor to some of the lab work they’ve been doing in my and Yanyan’s lab, respectively, as well as in collaboration with Brigitte Lavoie at UVM, with whom we have worked for several years on mouse models of inflammation.
Ashley has an extensive background in nutrition and dietetics (bio is below), and joined our labs to understand how dietary metabolites and gut microbiome metabolites could be used to improve health, and reduce inflammation in the gut and the brain (neuroinflammation). She is a Doctor of Philosophy candidate in Human Nutrition and Food Sciences, and previously completed her undergraduate degree in Food Science and Human Nutrition in 2021 as a Maine Top Scholar, her Master’s degree in Food Science and Human Nutrition with Jade McNamara, at UMaine on intuitive eating in college students, and completed a dietetic internship and shortly after passed her RD exam to become a registered dietitian in 2023. She is incredibly interested in nutrition therapy and is beginning her research looking into the microbiome and metabolomic pathways in the context of IBD.
Ethan Glenn is a master’s student at SUNY Binghamton University School of Pharmacy in Yanyan Li’s Lab. Ethan has been helping to develop several models of neuroinflammation to use for testing the efficacy of different preparations of broccoli sprouts in delivering bioactives.
Ashley and Ethan have been collaborating on protocol design and testing, sample processing, and data analysis for several projects, including one we recently completed which will form the basis of Ashley’s dissertation.
Johanna, Ashley, Alexis, and Sue put in long hours during the mouse trial to collect samples each day.
1 School of Food and Agriculture, University of Maine, Orono, Maine, USA 04469
2 School of Pharmacy and Pharmaceutical Sciences, SUNY Binghamton University, Binghamton, New York, USA 13902
3 Dept. of Neurological Sciences, University of Vermont College of Medicine, Burlington, Vermont, USA 05405
Abstract
Diet is increasingly recognized as a potential upstream modulator of the gut-brain axis (GBA) through its effects on the microbiome, microbial metabolites, and host immune and endocrine responses. The GBA is a complex, bidirectional network connecting the gastrointestinal tract and central nervous system, with diet influencing microbial community structure and metabolic output. Plant-based diets, such as Mediterranean and MIND, have been associated with increased production of anti-inflammatory microbial metabolites and improved barrier function, while high calorie/low nutrient diets are often linked to increased immune activation and barrier dysfunction. However, while microbial metabolites, especially short-chain fatty acids, indoles, bile acids, and isothiocyanates, have been proposed as mediators of neuroprotective effects, their role in neurodegenerative diseases remains an area of active investigation, with evidence largely derived from preclinical and associative human studies. Cruciferous vegetables, especially broccoli sprouts, are an emerging focus of research for their bioactive compound sulforaphane, which activates Nrf2-centered cytoprotective pathways. Animal and early human studies suggest sulforaphane can improve cognitive and behavioral outcomes, though larger clinical trials are needed. Personalized, microbiota-targeted dietary interventions may offer scalable strategies for managing neuroinflammatory and neurodegenerative conditions, and we emphasize the need for integrated research across diet, microbiome, and brain health.
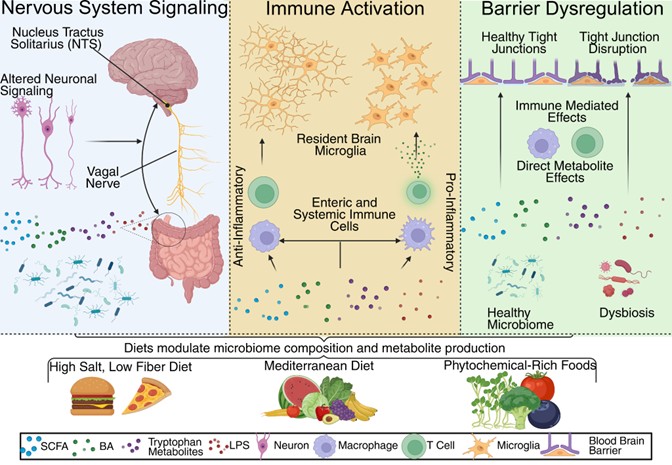
Figure 1: Mechanisms of how Diet Influences the Gut Brain Axis. Depicting three mechanisms of the gut brain axis of importance for dietary interventions. Different types of diets can modulate the microbiome to produce different metabolites. These metabolites can affect nervous system signaling through the ENS and vagal nerve. Immune activation via these metabolites either enterically or systemically can lead to activation of resident brain microglia. Dietary metabolites have also been shown to either protect blood brain barrier tight junctions or disrupt them, both through direct effects and immune mediated effects. Made in Biorender under license by E. Glenn.
The Lab is delighted to finally announce the publication of a study on the efficacy of a glucoraphanin supplement in converting to sulforaphane, and of the effect on or participation of gut microbes! The article can be found online here.
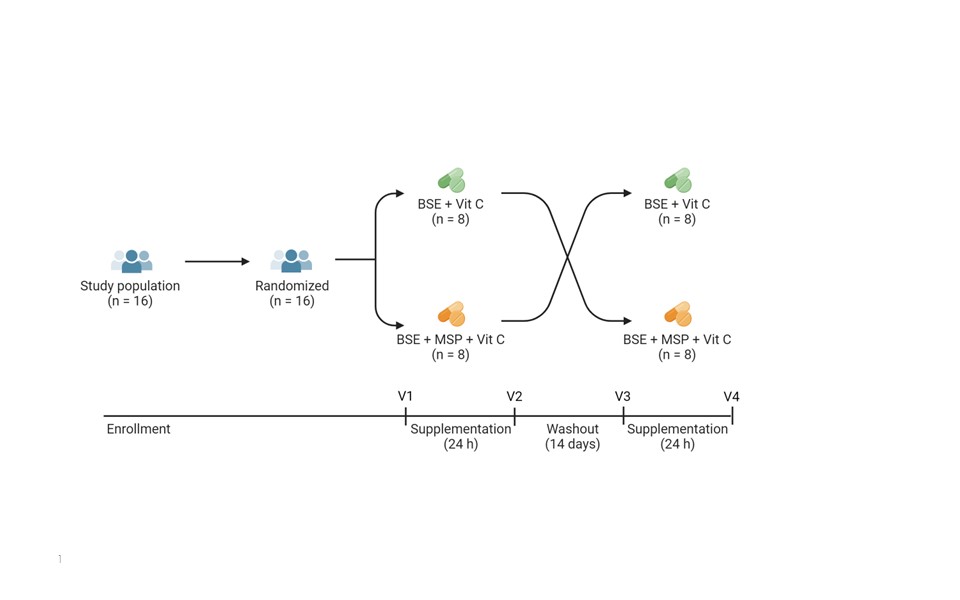
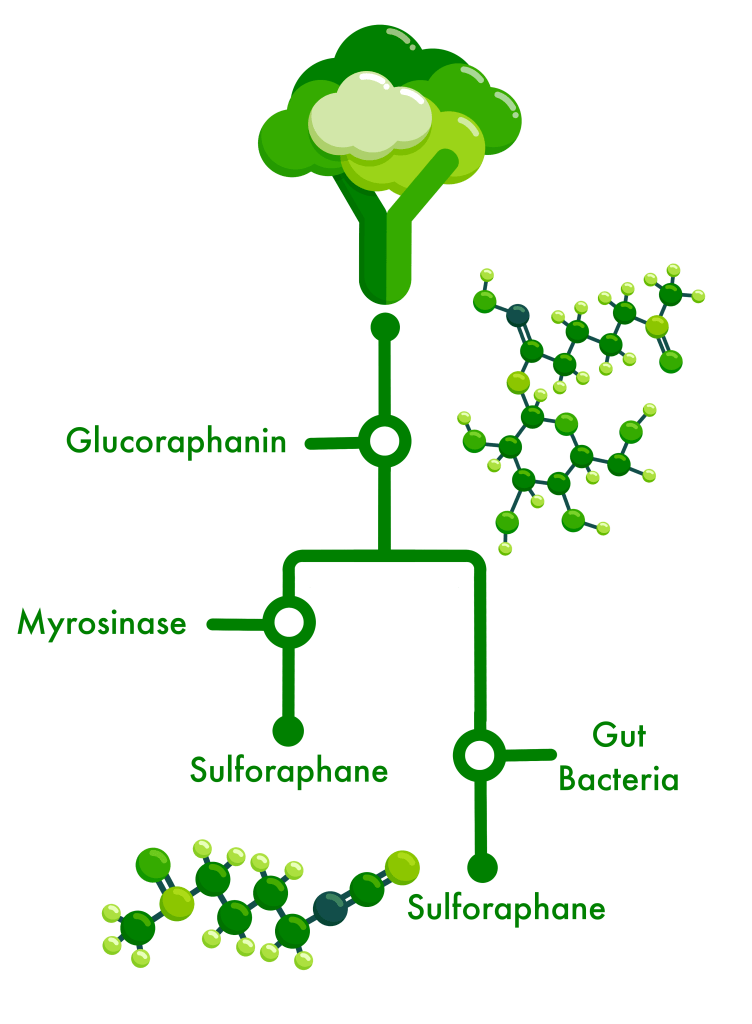
The study was run back in 2021 at Appalachian State University by Dr. Giuseppe Valacchi, Alessandra Pecorelli, and colleagues, when participants were given one dose of a glucoraphanin supplement combined with the plant enzyme myrosinase (which converts the GLR into the anti-inflammatory sulforaphane by the time the supplement gets to your small intestines), or one dose of the glucoraphanin supplement alone (in the absence of the plant enzyme, this requires gut microbes to convert GLR to SFN).
Yanyan and I were introduced to Dr. Jed Fahey when he joined the UMaine Institute of Medicine faculty network in 2022. We talked to Jed about his recent work Brassica Protection Products, a company he founded back in 1997 with Dr. Paul Talalay, and Yanyan and I were invited as collaborators in 2023 by Antony Talalay, CEO and Co-Founder, and Paul’s son.
As part of their dissertations, Marissa Kinney performed qPCR to quantify microbial genes during her master’s, and Lola Holcomb performed 16S rRNA bacterial community sequencing during her PhD. We submitted the manuscript for peer review in Dec 2024. After a slow review process, the paper was finally released in Feb 2026, and we’ve begun brainstorming the next step in our research. While Jed, Tony, and the BPP team are, of course, interested in how their supplement can be used to improve health, the Ishaq Lab is also interested in teasing apart why some people’s gut microbiome is very responsive to GLR supplementation and will produce a fair amount of sulforaphane, while other’s people’s gut won’t react to glucoraphanin at all.
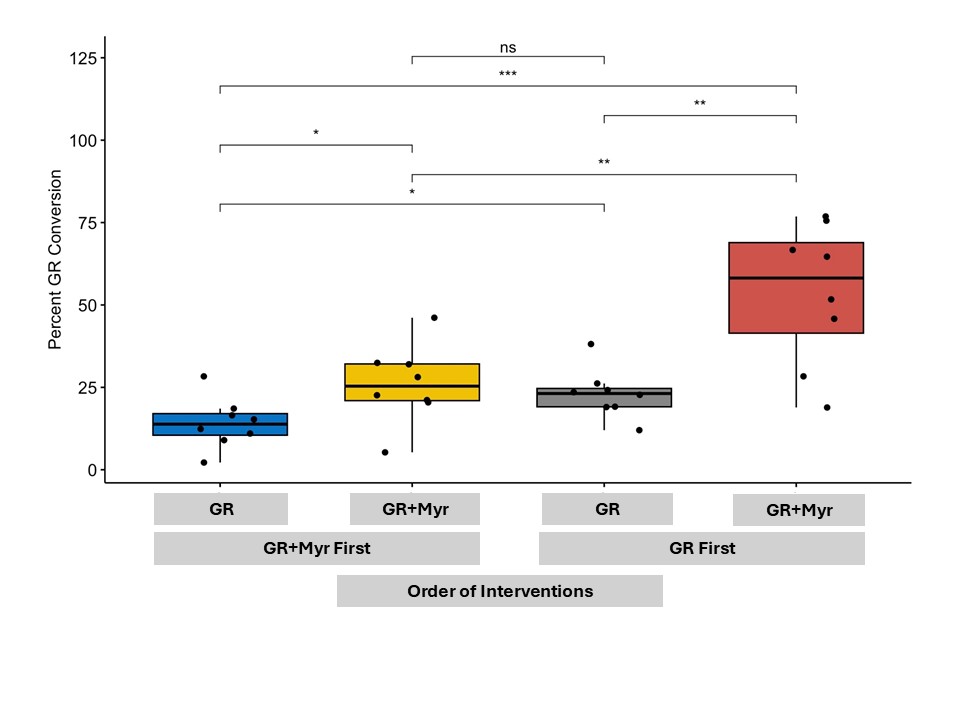
Even more intriguing were the participants in the study who had very little glucoraphanin conversion even when the myrosinase enzyme that can do the conversion was provided in the supplement. In fact, there was a lot of variability in how effective the enzyme was depending on the person- implying there are other biological or environmental factors at play which may be impeding the conversion of glucoraphanin into an anti-inflammatory.
Angela Mastaloudis, Lola Holcomb, Jed W. Fahey, Camila Olson, David C. Nieman, Colin Kay, Robert O’Donnell, Alessandra Pecorelli, Marissa Kinney, Yanyan Li, Suzanne L. Ishaq & Giuseppe Valacchi. Scientific Reports, In Press. (2026)
Abstract: Inactive glucoraphanin (GR) in broccoli is converted to the antioxidant, anti-inflammatory, and anti-bacterial sulforaphane (SF) by cruciferous vegetable enzyme myrosinase (Myr), or similar enzymes from specific gut bacteria; both sources have variable efficiency. The effects of exogenous Myr on the conversion efficiency of GR to SF was compared to gut microbial Myr-like activity. In a randomized, double-blind, crossover study, sixteen subjects (9 F: 7 M) received a single oral dose of GR in broccoli seed extract with Myr-containing mustard seed powder, or broccoli seed extract alone, both with ascorbic acid. GR + Myr, on average, doubled the bioavailability of SF (39.8 ± 3.1%) compared to GR alone (18.6 ± 3.1%), and increased the conversion rate in the first 8 h (25.4% ± 2.7%) compared to GR alone (8.0% ± 2.7) based on measurement of urinary metabolites. There were no differences in fecal bacterial communities after the single dose; however, four bacterial GR-converting genes significantly correlated with GR conversion (p < 0.0155). To our knowledge, this is the first human study to simultaneously investigate (1) a well-defined Myr source, (2) broccoli seeds as source of GR, (3) prediction of gut microbial responsiveness to GR.
The Ishaq Lab is testing out some probiotics in mice this spring, as we look towards the next phase of our broccoli sprouts and gut microbiome work: generating solutions. It has been five years since our last mouse experiments, in part, because we have been busy digging into samples, data, and ideas from those studies.
We gained valuable insight into which and how gut bacteria might metabolize the inactive glucosinolates from broccoli sprouts – glucoraphanin being the one we focus on – and produce byproducts like sulforaphane which our gut cells can use to reduce the chemical and physical damage caused by inflammation or oxidation.
This process is easy in the lab and tricky in the real world – not everyone has the bacteria in their gut which can do it, some have the bacteria but they are not active, and some have the bacteria but they are making a different version of the byproduct which we cannot use.
For the past three years, we have been screening >300 gut bacteria to identify and select ones with the ability to grow in the presence of GLR and metabolize it into the byproducts SFN or SFN-nitrile (which can be antimicrobial towards some bacteria).
In culture, the byproducts more-or-less all help the bacteria to survive by providing sulfur, glucose, or other chemicals it needs, but also by acting as an antioxidant compound that binds to a free oxygen (reactive oxygen species) so it doesn’t try to bond with chemicals on the surface of cells and cause them damage (oxidation damage). The byproducts also appear to help the bacteria thrive in acidic culture media which they otherwise can’t survive. When cultured with colon cells and GLR, some of those bacteria and their byproducts reduce inflammation.
The culmination of the past few years of work was to choose two bacterial cultures to try out our idea: that probiotics plus the broccoli sprout diet would help individuals who were not responsive to the diet. Because the gut microbiome, health, and the way that an individual and their microbes respond to diet are all very complicated processes that are specific to each person, it’s easier to test some of these concepts first in the lab or, which we did in early January, in animal models that mimic disease conditions in humans.
We ran a short trial in mice to find out if our two bacterial cultures that were so successful in the lab would also be effective when put back into the chaotic and competitive ecosystem of the gut. To further challenge our bacteria, we tested their ability to survive and reduce inflammation during a flare up of ulcerative colitis.
Every day for almost two weeks, we weighed each mouse (shown below) to make sure the colitis did not cause too much weight loss. Sue, Alexis, Johanna, and Ashley were all approved to handle the mice, so we were in charge of picking them up to weigh them. This was no easy task – mice are agile!
Each day, we also collected feces from the cages to check for diarrhea, or for blood, which are two symptoms of colitis. Our undergrads Madison and Brian worked tirelessly to tweeze feces into collection tubes, and to use the FOBT cards to check for blood.
We used custom made gelatin cubes filled with probiotic to deliver our treatments. The gelatin stuck to the side of the cages which allowed us to easily see that our mice were consuming their probiotic.
It will take us months to process some of the samples we collected which are the most cost-effective to run, and the rest will have to wait in the freezer until we receive more funding (which could take months or years as the changes to the federal funding system have doubled the time it takes for proposals to be reviewed and the ~5-20% of accepted projects to receive funding). We collected >500 fecal samples (each with 5 – 10 pellets/sample), 200 gut samples, 100 intestinal tissue samples, and 50 blood samples! To help maximize the benefits of this experiment and use all parts of these mice we also collected samples for a course at UMaine which teaches pre-medical, pre-veterinary, nursing, biology, and other health-focused students how to make and read tissue slides, to better understand anatomy, physiology, developmental biology, and health.
Still, we gained valuable data already, and the experiment provided a unique opportunity for students to receive hands-on-training for evaluating disease intensity using fecal samples, using tissues to make slides for histology, evaluating intestinal damage to tissues, collecting samples using aseptic technique to prevent contaminating them, working safely with microbes, and collaboratively working as a team to advance knowledge of health. Myself and our grad team (Johanna, Alexis, and Ashley) managed the project, and our undergrad team (Madison, Brian, Aaron, and Alexandra) were there to help us label ~1000 tubes for sample collection, and collect hundreds of fecal pellets out of the used bedding so we could track mouse microbes. Undergrads were also able to learn some general mouse care and research facility care from the ‘mouse house’ technician at UMaine, Alexis R. A former UMaine undergrad in the AVS program, Alexis R. manages and cares for a wide variety of animal species and she was instrumental in helping us manage our intensive sample collection schedule.
Johanna, Ashley, Alexis, and Sue put in long hours during the mouse trial to collect samples each day.
This project was made possible by the help of many. Again, we are grateful to Alexis and the UMaine CORE staff who not only support research at specialized facilities but helped us to afford to run our pilot project, to Emma who runs the UMaine Electron Microscopy Lab for teaching students histology and microscopy, to all the undergraduate and graduate students who worked tirelessly to help each other on this project, and to the funding agencies which supporting the lab work that helped us get to this project: the Biomedical Association of Maine (graduate awards), the Crohn’s and Colitis Foundation (graduate awards), the NIH NIDDK, and the USDA.
Since the summer of 2023, I have been part of an interdisciplinary team that examines the way microbiome researchers use social and population descriptors for people in their analysis. In many cases, only basic information about a person is available in large datasets that are publicly available to use, or detailed information about a person is difficult to obtain during a study, thus many researchers rely on “proxy terms” to try and understand how human microbiomes are assembled and changed. Proxy terms are broad categories that group people, such as geographic area or race, but often these are too broad to be used for any meaningful analysis, especially when working with biological data.
‘Race’ is a relatively new concept used to describe social groups, and as discussed brilliantly in the National Academies of Science, Engineering, and Medicine’s report on “Use of Race, Ethnicity, and Ancestry as Population Descriptors in Genomics Research“, it has been mis-used for several hundred years to insinuate basic biological differences between people. This was done intentionally to justify discrimination all the way up to slavery, but it has been unintentionally propagated into research through the use of race as a proxy term to represent someone’s lifestyle. In recent decades, microbiome research has been trying to understand how human lives affect the microbiomes they accumulate, and similarly has sometimes incorrectly espoused the idea that vague social categories manifest as biological differences.
Our group delved in the history of race in biological science, case studies where results that implicate race led to discriminatory policy and practice, and give guidelines for selecting more specific factors to understand the social and environmental impacts on the microbiome.
Authors: Nicole M. Farmer1,2, Amber Benezra1,3, Katherine A. Maki1, Suzanne L. Ishaq1,2,Ariangela J. Kozik1,2,4,5*
Affiliations:
1 The Microbes and Social Equity working group, Orono, Maine, USA; 2 Nova Institute for Health, Baltimore, MD; 3 Science and Technology Studies, Stevens Institute of Technology, Hoboken, New Jersey, USA; 4 Division of Pulmonary and Critical Care Medicine, Department of Internal Medicine, University of Michigan, Ann Arbor, Michigan, USA; 5 Department of Molecular, Cellular, and Developmental Biology, University of Michigan, Ann Arbor, Michigan, USA
Abstract
Microbiome science is a celebration of the connections between humans, our environment, and microbial organisms. We are continually learning more about our microbial fingerprint, how each microbiome may respond to identical stimuli differently, and how the quality of the environmental conditions around us influences the microorganisms we encounter and acquire. However, in this process of self-discovery, we have utilized socially constructed ideas about ourselves as biological factors, potentially obscuring the true nature of our relationships to each other, microbes, and the planet. The concept of race, which has continuously changing definitions over hundreds of years, is frequently operationalized as a proxy for biological variation and suggested to have a real impact on the microbiome. Scientists across disciplines and through decades of research have misused race as a biological determinant, resulting in falsely scientific justifications for social and political discrimination. However, concepts of race and ethnicity are highly nuanced, inconsistent, and culturally specific. Without training, microbiome researchers risk continuing to misconstrue these concepts as fixed biological factors that have direct impacts on our microbiomes and/or health. In 2023, the National Academies of Sciences, Engineering, and Medicine released recommendations on the use of population descriptors such as race and ethnicity in genetic science. In this paper, we posit similar recommendations that can and must be translated into microbiome science to avoid re-biologizing race and that push us toward the goal of understanding the microbiome as an engine of adaptation to help us thrive in a dynamic world.
Heather Richard passed her comprehensive exam, which means she is advancing to PhD candidacy!! Heather’s research focuses on how land use and water infrastrucure changes the dynamics of salt marshes and their tidal creeks, which alters their microbial communities, biochemical processes, and capacity to sequester atmospheric carbon in sediment. She details that work, including field sampling and labwork protocols, as well as data visualization and major findings, on an interactive website she created to support research into salt marshes in Maine.
Heather Richard, B.A., M.S.
Doctor of Philosophy Candidate, Ecology and Environmental Sciences. Heather is being co-advised by Dr. Peter Avis
Heather joined the University of Maine in 2021 as a PhD student with the Maine eDNA program and studies the impacts of bridges and roads on microbial communities in salt marsh habitats. Her background in Ecology led her to pursue a career in informal environmental education for several years before getting a Master’s degree in Marine Biology from San Francisco State University studying biofilms on microplastics pollution. Upon returning to Maine in 2016 she led local research for a coastal non-profit organization and has since been dedicated to studying coastal environmental issues relevant to Maine. She has found a true passion in bioinformatic analysis and is eager to learn new tools for data analysis of all kinds.
The exam involved writing mini-papers around topics assigned by her committee, including decision-making and troubleshooting for sequencing data analysis, biogeochemical processes in salt marshes that drive carbon sequestration or release, and microbial ecology in coastal ecosystems. Each set of questions were released once a day for 5 days, and mini-papers took 6 -8 hours to complete and had to be returned within 24 hours. This intensive series of written exams were followed by a two-hour question-and-answer session in which Heather gave further detail on her written answers, connected basic biochemical processes to broader ecosystem-level microbial ecology, and considered furture research designs. This grueling process is the last hurdle for PhD students, and now it’s “smooth sailing” until the PhD defense. Over the next year or so, Heather will perform metegenomics sequencing data analysis from her salt marsh sites, and synthesizing microbial and biochemical data into several manuscripts which we will submit to scientific journals for peer review, and eventual publication.
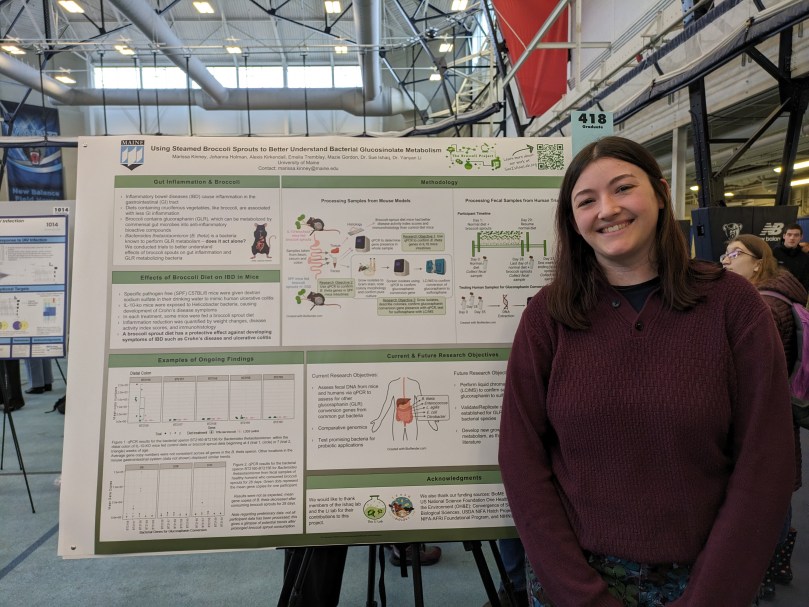
Lab rockstar, Marissa Kinney, successfully defended her master’s thesis today!!! After a well-earned holiday break, Master Kinney is pursuing a career in biomedical/microbiological research.
Marissa was Master of Science student in Microbiology, and a researcher in the One Health and the Environment program, both of which are prestigious graduate programs at UMaine, from Jan 2023 – Dec 2024. She loves learning and bench microbiology, and she employed these passions on multiple lab projects investigating the bacteria which transform glucoraphanin in broccoli sprouts into the anti-inflammatory sulforaphane in the gut. The focus of her time has been to develop new lab protocols, refine existing ones and make them easier for new lab members to learn, and to share her expertise by teaching other students in the lab. She’s excelled at these objectives so well, that in the past two years many people assumed she was a Lab Manager rather than a student.
Marissa has been extremely productive in the last two years: in her first three months she contributed lab work to two publications on broccoli sprout diets in mouse models of Inflammation Bowel Disease in 2023, and has since contributed to another manuscript currently in review on glucoraphanin supplements and gut microbiome changes in people, and two more manuscripts in preparation on culturing gut microbiota, and a broccoli sprout diet in people. It’s no surprise that Marissa has been an author on so many papers in so little time — she led a publication when she was an undergraduate! You can check her Google Scholar page for more info on these papers. Marissa has also presented this work on campus at the UMaine Student Research Symposium twice, as well as attended conferences for the American Society for Nutrition and the American Society for Microbiology for professional development.
Marissa Kinney presenting research.
The Ishaq Lab at Boda Borg in Boston, summer 2023. We are showing how many quests we completed.
Lab lunch with Mira Talalay, Sue, and Marissa Kinney
Previous to being in the lab, Marissa completed her undergraduate at the University of Maine in 2021, earning a BS in Microbiology and a BS in Cellular/Molecular Biology. She devoted a large portion of her time in undergrad to research in the laboratories of Dr. Julie Gosse and Dr. Edward Bernard. After graduating, she worked in the field of public health at UMaine’s Margaret Chase Smith Policy Center, collecting and processing data about violent and drug-related deaths in Maine. While her role at the Center was one she loved dearly, she felt a big pull towards laboratory work and academic research, and her graduate work enforced this passion. Marissa has been a core member of the lab, and we’ll miss her!! She plans to pursue a research career here in Maine after defending and enjoying a well-earned vacation.
USING BROCCOLI SPROUT DIETS TO UNDERSTAND GUT BACTERIAL GLUCOSINOLATE METABOLISM TO RESOLVE INFLAMMATORY BOWEL DISEASE
Abstract
Globally, millions of people have been diagnosed with a type of inflammatory bowel disease (IBD). These diseases cause dysfunction of the gastrointestinal (GI) tract, resulting in a wide range of symptoms that create a disruption in overall health. Research has suggested that diet and the microbial community composition of the gut microbiome play a significant role in regulating gastrointestinal inflammation. Specifically, studies have shown that diets high in cruciferous vegetables, such as broccoli, are associated with a reduction in gastrointestinal inflammation. Glucoraphanin is a compound present in broccoli that can be metabolized by gut bacteria to become an anti-inflammatory compound known as sulforaphane. Our initial research showed that the administration of a broccoli sprout diet to mouse models for Crohn’s disease and ulcerative colitis, two major types of IBD, yields inflammation reduction and symptom resolution. For these trials, fecal samples obtained from different sections of the mouse bowel were tested for presence of glucoraphanin-metabolizing genes present in a common gut bacteria, Bacteroides thetaiotaomicron (B. theta). Glucoraphanin conversion is higher and more reliable in mice than in people, however mouse models are not perfect representatives of humans. Hoping to understand the impacts of broccoli sprouts on the human gut microbiome, fecal samples were obtained from healthy individuals who consumed broccoli sprouts for 28 consecutive days, as long-term diet interventions are needed to meaningfully change gut microbial communities. In a separate trial conducted by the scientists at Brassica Protections Product, fecal samples were collected from people who were administered a single dietary supplement containing a high dose of glucoraphanin with and without plant-sourced myrosinase, as a means of evaluating the effectiveness of glucoraphanin conversation which was or was not reliant on gut microbiota, respectively. These samples were analyzed for glucoraphanin metabolizing genes from B. theta and other commensal gut bacteria. Data collected from these human trial experiments aided in understanding the impacts of a whole food broccoli sprout diet and supplementation of glucoraphanin on the bacterial community composition of the gut microbiota. Additionally, this work will help grow and strengthen the current knowledge on broccoli as an anti-inflammatory and the variabilities present in the gut microbiomes of humans.
Lab rockstar, Marissa Kinney, is set to defend her Master’s of Science thesis in Microbiology on Wednesday, December 18th!! The thesis abstract is below.
You can register here for the publicly available seminar portion from 1 – 2 pm ET on Zoom
Marissa has been a Master of Science student in Microbiology, and a researcher in the One Health and the Environment program, both of which are prestigious graduate programs at UMaine, for the last two years. She loves learning and bench microbiology, and she employed these passions on multiple lab projects investigating the bacteria which transform glucoraphanin in broccoli sprouts into the anti-inflammatory sulforaphane in the gut. The focus of her time has been to develop new lab protocols, refine existing ones and make them easier for new lab members to learn, and to share her expertise by teaching other students in the lab. She’s excelled at these objectives so well, that in the past two years many people assumed she was a Lab Manager rather than a student.
Marissa has been extremely productive in the last two years: in her first three months she contributed lab work to two publications on broccoli sprout diets in mouse models of Inflammation Bowel Disease in 2023, and has since contributed to another manuscript currently in review on glucoraphanin supplements and gut microbiome changes in people, and two more manuscripts in preparation on culturing gut microbiota, and a broccoli sprout diet in people. It’s no surprise that Marissa has been an author on so many papers in so little time — she led a publication when she was an undergraduate! You can check her Google Scholar page for more info on these papers. Marissa has also presented this work on campus at the UMaine Student Research Symposium twice, as well as attended conferences for the American Society for Nutrition and the American Society for Microbiology for professional development.
Marissa Kinney presenting research.
The Ishaq Lab at Boda Borg in Boston, summer 2023. We are showing how many quests we completed.
Lab lunch with Mira Talalay, Sue, and Marissa Kinney
Previous to being in the lab, Marissa completed her undergraduate at the University of Maine in 2021, earning a BS in Microbiology and a BS in Cellular/Molecular Biology. She devoted a large portion of her time in undergrad to research in the laboratories of Dr. Julie Gosse and Dr. Edward Bernard. After graduating, she worked in the field of public health at UMaine’s Margaret Chase Smith Policy Center, collecting and processing data about violent and drug-related deaths in Maine. While her role at the Center was one she loved dearly, she felt a big pull towards laboratory work and academic research, and her graduate work enforced this passion. Marissa has been a core member of the lab, and we’ll miss her!! She plans to pursue a research career here in Maine after defending and enjoying a well-earned vacation.
USING BROCCOLI SPROUT DIETS TO UNDERSTAND GUT BACTERIAL GLUCOSINOLATE METABOLISM TO RESOLVE INFLAMMATORY BOWEL DISEASE
Abstract
Globally, millions of people have been diagnosed with a type of inflammatory bowel disease (IBD). These diseases cause dysfunction of the gastrointestinal (GI) tract, resulting in a wide range of symptoms that create a disruption in overall health. Research has suggested that diet and the microbial community composition of the gut microbiome play a significant role in regulating gastrointestinal inflammation. Specifically, studies have shown that diets high in cruciferous vegetables, such as broccoli, are associated with a reduction in gastrointestinal inflammation. Glucoraphanin is a compound present in broccoli that can be metabolized by gut bacteria to become an anti-inflammatory compound known as sulforaphane. Our initial research showed that the administration of a broccoli sprout diet to mouse models for Crohn’s disease and ulcerative colitis, two major types of IBD, yields inflammation reduction and symptom resolution. For these trials, fecal samples obtained from different sections of the mouse bowel were tested for presence of glucoraphanin-metabolizing genes present in a common gut bacteria, Bacteroides thetaiotaomicron (B. theta). Glucoraphanin conversion is higher and more reliable in mice than in people, however mouse models are not perfect representatives of humans. Hoping to understand the impacts of broccoli sprouts on the human gut microbiome, fecal samples were obtained from healthy individuals who consumed broccoli sprouts for 28 consecutive days, as long-term diet interventions are needed to meaningfully change gut microbial communities. In a separate trial conducted by the scientists at Brassica Protections Product, fecal samples were collected from people who were administered a single dietary supplement containing a high dose of glucoraphanin with and without plant-sourced myrosinase, as a means of evaluating the effectiveness of glucoraphanin conversation which was or was not reliant on gut microbiota, respectively. These samples were analyzed for glucoraphanin metabolizing genes from B. theta and other commensal gut bacteria. Data collected from these human trial experiments aided in understanding the impacts of a whole food broccoli sprout diet and supplementation of glucoraphanin on the bacterial community composition of the gut microbiota. Additionally, this work will help grow and strengthen the current knowledge on broccoli as an anti-inflammatory and the variabilities present in the gut microbiomes of humans.
The new fall semester always brings new undergraduate researchers to the lab, and we are pleased to welcome these students to #TeamBroccoli! A few other undergraduates who did not contibute bios yet have also been shadowing in the lab this semester, and are considering joining the lab to complete their senior research projects.
New undergrad researchers joined us in 2024 from several programs on campus!
Isaac Mains
Undergraduate Researcher, Microbiology
Isaac is a BS microbiology student working to complete his undergraduate degree at the University of Maine in 2025. After graduation, he plans to pursue a Masters’ in medical science and ultimately wishes to attend medical school to become a practicing physician. His research interests include the gut microbiome and dysbiosis. He grew up in Bar Harbor, Maine, and has a great appreciation for the outdoors, stemming from his years spent living in Acadia. He joined the Ishaq lab in fall 2024, and is looking forward to helping spread awareness of issues surrounding social equity and host microbiome interactions.
Miriam Talalay
Undergraduate researcher, Zoology and Veterinary Studies Mira is in her third year at University of Maine where she is studying Zoology and Veterinary Studies with the goal of becoming a wildlife researcher and rehabilitator. She is from Maryland but grew up spending time at her grandparents’ camp in Surry, Maine. Her grandfather (Dr. Paul Talalay) is the researcher from Johns Hopkins University who discovered the chemoprotective properties of sulforaphane from broccoli, so she is happy to be a part of his legacy on Team Broccoli.
Mira is an avid wildlife photographer and kayaker. She also plays violin in her spare time and has played with the University of Maine orchestra. Prior to coming to UMaine, she was awarded Duke of Edinburgh’s International Silver Award USA , Young Woman of the Year (Baltimore County Commission for Women), President’s Volunteer Service Award, US Congressional Award for Voluntary Service and Personal Development (Gold Medal), and a Maryland Governor’s Citation for Voluntarism. She is currently working on the Gold Medal requirements for the Duke of Edinburgh’s International Award.
Emelia Tremblay
Undergraduate Researcher, Microbiology, University of Maine
I am an undergraduate who is due to graduate in the Spring of 2025. Outside of the lab I work in Athletics as a Student Supervisor, I am the Vice President of Academics in Delta Phi Epsilon, and the Vice President of Judicial Affairs and Risk Management of the Panhellenic Council at UMaine. I was a Quality Intern in the laboratory at Edesia Nutrition, which partners with UNICEF and WFP to provide a fortified peanut butter supplement for malnourished babies in underdeveloped countries. I hope to attend grad school to further my education in either microbiology or public health. I would like to ultimately work in a research lab studying the human microbiome or work to bridge the gap between scientists and the general public through Public Health Communication.
Grad Students
Heather Richard is a PhD student who has been working with the lab for over a year on her project, and who formally joined the lab in early 2024. Since her field work is seasonal, it took us awhile to get her welcome post put together!
Heather Richard, B.A., M.S.
Doctor of Philosophy student, Ecology and Environmental Sciences. Heather is being co-advised by Dr. Peter Avis
Heather joined the University of Maine in 2021 as a PhD student with the Maine eDNA program and studies the impacts of bridges and roads on microbial communities in salt marsh habitats. Her background in Ecology led her to pursue a career in informal environmental education for several years before getting a Master’s degree in Marine Biology from San Francisco State University studying biofilms on microplastics pollution. Upon returning to Maine in 2016 she led local research for a coastal non-profit organization and has since been dedicated to studying coastal environmental issues relevant to Maine. She has found a true passion in bioinformatic analysis and is eager to learn new tools for data analysis of all kinds.
A scientific article led by my colleague Dr. Alaa Rabee at the Desert Research Center in Egypt was just published online and is now available! Dr. Rabee and I have been collaborating remotely on projects related to the bacteria in the rumen of camels, sheep, and cows, as Dr. Rabee’s work focuses on the isolation of bacteria which can degrade plant materials efficiently and could be used to produce biofuels. He will be spending 6 months working in my lab as a visiting scholar, which was delayed until this year because of the pandemic.
Rabee, A.E., Sayed Alahl, A.A., Lamara, M., Ishaq, S.L. 2022. Fibrolytic rumen bacteria of camel and sheep and their applications in the bioconversion of barley straw to soluble sugars for biofuel production. PLoS ONE 17(1): e0262304. Article.
Abstract
Lignocellulosic biomass such as barley straw is a renewable and sustainable alternative to traditional feeds and could be used as bioenergy sources; however, low hydrolysis rate reduces the fermentation efficiency. Understanding the degradation and colonization of barley straw by rumen bacteria is the key step to improve the utilization of barley straw in animal feeding or biofuel production. This study evaluated the hydrolysis of barley straw as a result of the inoculation by rumen fluid of camel and sheep. Ground barley straw was incubated anaerobically with rumen inocula from three fistulated camels (FC) and three fistulated sheep (FR) for a period of 72 h. The source of rumen inoculum did not affect the disappearance of dry matter (DMD), neutral detergent fiber (NDFD). Group FR showed higher production of glucose, xylose, and gas; while higher ethanol production was associated with cellulosic hydrolysates obtained from FC group. The diversity and structure of bacterial communities attached to barley straw was investigated by Illumina Mi-Seq sequencing of V4-V5 region of 16S rRNA genes. The bacterial community was dominated by phylum Firmicutes and Bacteroidetes. The dominant genera were RC9_gut_group, Ruminococcus, Saccharofermentans, Butyrivibrio, Succiniclasticum, Selenomonas, and Streptococcus, indicating the important role of these genera in lignocellulose fermentation in the rumen. Group FR showed higher RC9_gut_group and group FC revealed higher Ruminococcus, Saccharofermentans, and Butyrivibrio. Higher enzymes activities (cellulase and xylanase) were associated with group FC. Thus, bacterial communities in camel and sheep have a great potential to improve the utilization lignocellulosic material in animal feeding and the production of biofuel and enzymes.
Funded by the University of Maine Rural Health and Wellbeing Grand Challenge Grant Program, this project assesses pathogen carriage by mice and flying squirrels on or near farms in several locations in Maine. We live-capture mice and flying squirrels in traps, collect the poop they’ve left in the trap, and conduct a few other health screening tests in the field before releasing them. To maximize the information we collect while minimizing stress and interference to the animals, information is being collected for other projects in the Levesque Lab at the same time. We will be collecting samples for another few weeks, and then working on the samples we collected in the lab over the fall and winter.
One of the major goals of the funding program, and this project, is to engage students in research. After a few months on the project, some of our students describe their role and their experiences so far…
Marissa Edwards
Undergraduate in Biology
Levesque Lab
Hi! My name is Marissa Edwards and I am an undergraduate research assistant with Danielle Levesque. This summer, my role has been to set traps, handle small mammals, and collect fecal and tissue samples from deer mice.
One of the skills I’ve learned this summer is how to properly ear tag a mouse. To catch mice, we set traps across UMaine’s campus as well as other parts of Maine, including Moosehead Lake, Flagstaff Lake, and Presque Isle.
During our trip to Moosehead Lake, I saw a marten for the first time (it was in one of our traps). I did not know martens existed and initially thought it was a fisher cat. It was both a cool and terrifying experience!
Elise Gudde
Master’s Student of Ecology and Environmental Sciences
Levesque Lab
Hello, my name is Elise Gudde, and I am currently a master’s student at the University of Maine in the Ecology and Environmental Sciences program. I work in Dr. Danielle Levesque’s lab studying small mammal physiology in Maine.
This summer, as a part of the squirrel project, I work to trap small mammal species in Maine, such as white footed mice, deer mice, and flying squirrels in order to determine which species have shifted their range distributions as a result of climate change. Being a part of the research team, this summer has brought me all over Maine! I have been able to travel to Orono, Greenville, New Portland, and Aroostook County to study many interesting mammals. I even got to handle an Eastern chipmunk for the first time! As a member of the animal-handling side of the research team, I also collect fecal and tissue samples from the animals. These samples are then handed off for other members of the team to research in the lab!
Rebecca French
Undergraduate in Animal and Veterinary Sciences
Ishaq Lab
In the beginning of this project, I had no idea what I was getting myself into when I began researching flying squirrels and mice. I came into it with almost no in-person lab experience, so I had a lot to learn.
So far, I have been focusing on making media on petri dishes for culturing bacterial growth and after plating fecal bacteria on said plates; discerning what that growth can be identified as.
We are using media with specific nutrients, and colored dyes, and certain bacteria we are interested in will be able to survive or produce a color change. I have also been performing fecal flotations and viewing possible eggs and parasites under a microscope. What I’ve found most fun about this project is putting into practice what I have learned only in a classroom setting thus far. It is also very satisfying to be a part of every step of the project; from catching mice, to making media, to using that media to yield results and then to be able to have a large cache of information to turn it all into a full fledged project.
Some of the media used to culture bacteria from the feces of mice and squirrels.
Joe Beale
Undergraduate in Animal and Veterinary Sciences
Kamath Lab
Hello! My name is Joseph Beale, and I am an undergraduate at the University of Maine working on the squirrel project as a part of my capstone requirement for graduation. My primary responsibility in this project is the molecular testing of samples obtained from the field. Primarily I will be working with ear punch samples taken from flying squirrels and field mice. DNA extracts from these field samples will be run via qPCR. The results of this qPCR will tell us if these squirrels are carrying any pathogens.
The pathogens we will be testing for are those found in Ixodes ticks. The qPCR panel which we will be running the extracted DNA from the ear punches on tests for Borrelia burgdorferi, the causative agent of Lyme disease, Anaplasma phagocytophilum, the causative agent of anaplasmosis, and Babesia microti, the causative agent of Babesiosis. These pathogens and respective diseases discussed are all transmitted through Ixodes ticks. Deer ticks are the most common and famous of the Ixodes genus. The Ixodes genus encapsulates hard-bodied ticks. Along with deer ticks, Ixodes ticks found in Maine include: woodchuck ticks, squirrel ticks, mouse ticks, seabird ticks, and more. Mice and squirrel are ideal hosts for these Ixodes ticks, therefore becoming prime reservoirs for these diseases. In our research, we are interested in determining the prevalence of these diseases in squirrels and mice as these hosts can spread these diseases to humans and other animals in high tick areas.
qPCR, quantitative polymerase chain reaction, allows for the quantification of amplified DNA in samples. This will help tell us if these pathogens are present in samples and in what capacity. In qPCR provided DNA strands are added to the reaction. These strands match with the genome of the intended pathogens. If the pathogens are present in our samples, the provided DNA strands will bind to the present pathogen DNA. PCR will then work to manufacture billions of copies of this present pathogen DNA.
When not working on this project, I also work in the University of Maine Cooperative Extension Diagnostic Research Laboratory as a part of the Tick Lab. In this position I have honed the molecular biology skills that I will in turn use for the squirrel project.
Hello everyone! My name is Yvonne Booker and I am a rising senior, animal and poultry science major at Tuskegee University in Tuskegee, Alabama. I am interested in animal health research, with a particular focus in veterinary medicine. I’ve always wanted to be a veterinarian, but as I progressed throughout college, I became interested in learning more about animal health and how I might help animals on a much larger and impactful scale–which led me to the REU ANEW program. Currently climate change is causing an increase in global temperatures, putting pressure on animals’ ability to interact and survive within their environment. Consequently, scientists are now attempting to understand not just how to prevent climate change, but how these creatures are adapting to this emerging challenge.
My research experience this summer is geared toward addressing this global issue. I am currently working in Dr. Danielle Levesque’s Lab, which aims to study the evolutionary and ecological physiology of mammals in relation to climate. My project involves conducting a literature review of the microbiome of mammals, to learn more about how their microbial community plays a role in how they adapt in a heat-stressed environment.
Our knowledge of vertebrate-microbe interactions derives partly from research on ectotherms. While this research paves the path for a better understanding of how organisms react to temperature changes, fewer studies have focused on how mammals deal with these extreme temperature shifts—specifically, the abrupt surge in climate change. The ability of endotherms to thermoregulate alters our knowledge of (1) how mammals create heat tolerance against these environmental challenges and (2) how this internal process alters mammals’ adaptability and physiology over time. We suggest that the microbiome plays an essential part in understanding mammals’ heat tolerance and that this microbial community can help researchers further understand the various processes that allow mammals to survive extreme temperatures.
As a student of the REU ANEW program my goal was to go out of my comfort zone and study animals in an applied fashion that would impact animal health on an environmental and ecological scale; and this program was just that! My mentor, Dr. Levesque was wonderful in guiding me through conducting this research, while giving me the independence to create my own voice. The program directors, Dr. Anne Lichtenwalner and Dr. Kristina Cammen, have also been extremely supportive throughout this entire program equipping students with the tools they need to succeed as researchers. Although research was my primary focus this summer, some of my favorite memories involved building community with the students and the staff. From weekly check-ins on zoom to virtual game nights of complete smiles and laughter, this program has been one for the books! The One Health and the Environment approach to this Research Experience for Undergraduate students has encouraged me to build on my curiosity within the field of science, and I’m looking forward to applying what I’ve learned to my career in the future.