Scallops are a diverse animal group of marine bivalve mollusks (family Pectinidae) with global distribution in coastal waters, and Atlantic deep-sea scallops, Placopecten magellanicus, are found along the eastern coast of the United States and Canada. Scallops’ reproductive potential and industry demand make them a prime target for hatchery- and farm-based production, and this has been successfully achieved in bay scallops, but not in sea scallops. Currently, hatcheries collect wild sea scallop adults, or maintain cultured broodstocks, and spawn them in their facilities with the intention of forming a plentiful population to grow to adulthood, spawn, and sell to create a sustainable production cycle while also reducing disruption to the scallops’ natural habitat.

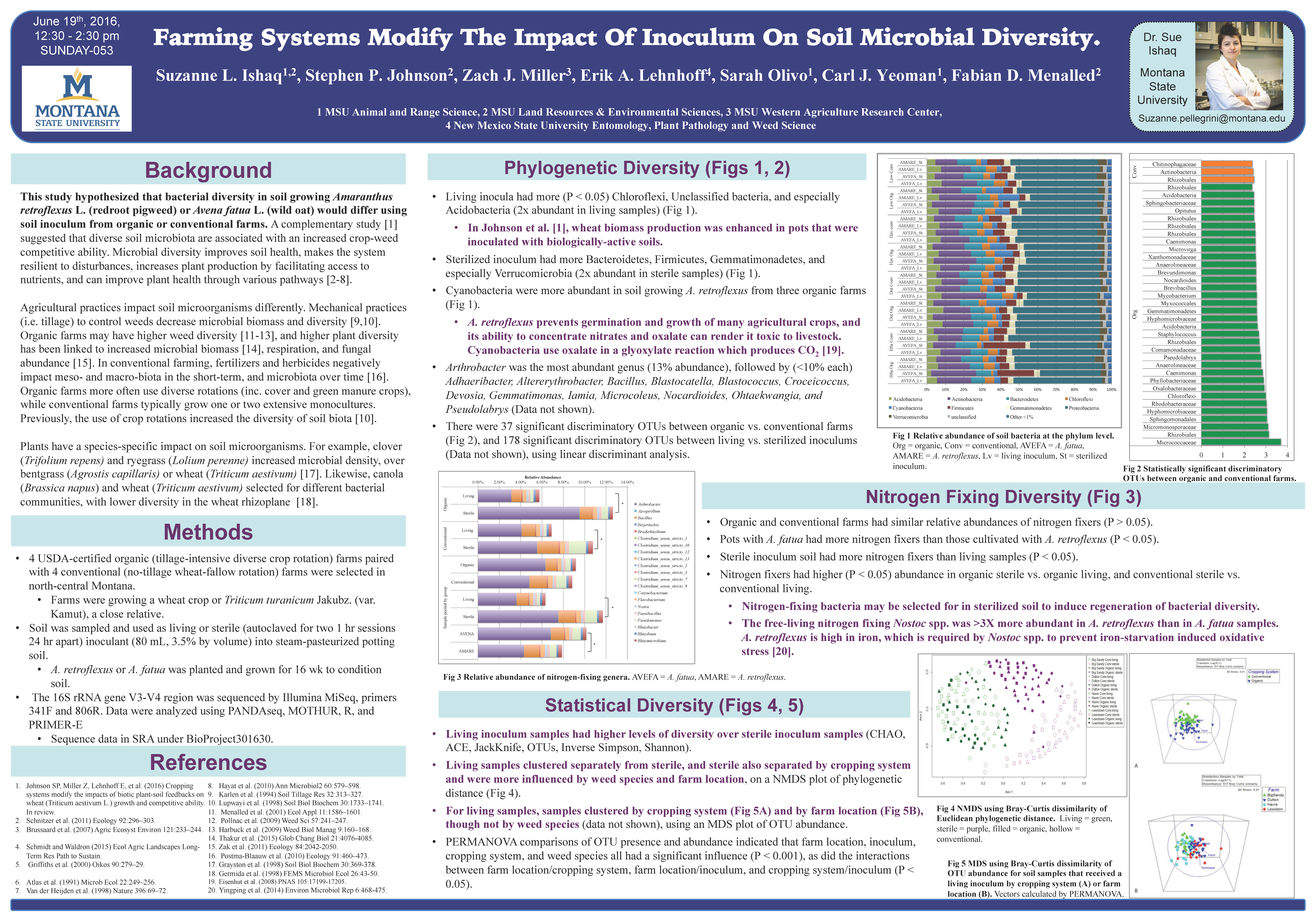
Unfortunately, in sea scallop hatcheries the last two weeks of the larval maturation phase, the veliger-stage, is plagued by large mortality events, going from 60 million sea scallop larvae down to several thousand individuals in a span of 48 hours. Survival of clutches to maturity remains very low, with an industry-standard rate around 1%. This drastic winnowing of larvae reduces the availability of cultured sea scallop spat for farmers, forcing sea scallop farms to rely almost exclusively on sea scallop spat collected from wild populations for stock and is seen as a bottleneck for growth of the industry and achieving sustainable harvests. Hatchery larval die-off is well-demonstrated not to be caused by inadequate diet, lighting, temperature, or atmospheric pressure in aquaculture facilities compared to wild conditions.
This project wanted to know if there was any clue in the bacteria that associate with larvae, or with the tanks they are in. In particular, hactcheries are worried about certain species of bacteria in the genus Vibrio, as they can cause disease to scallops and/or people, but it is tricky to study them because there are many species which do nothing at all. This project is part of another experiment to examine some of the Vibrio we found in tanks.
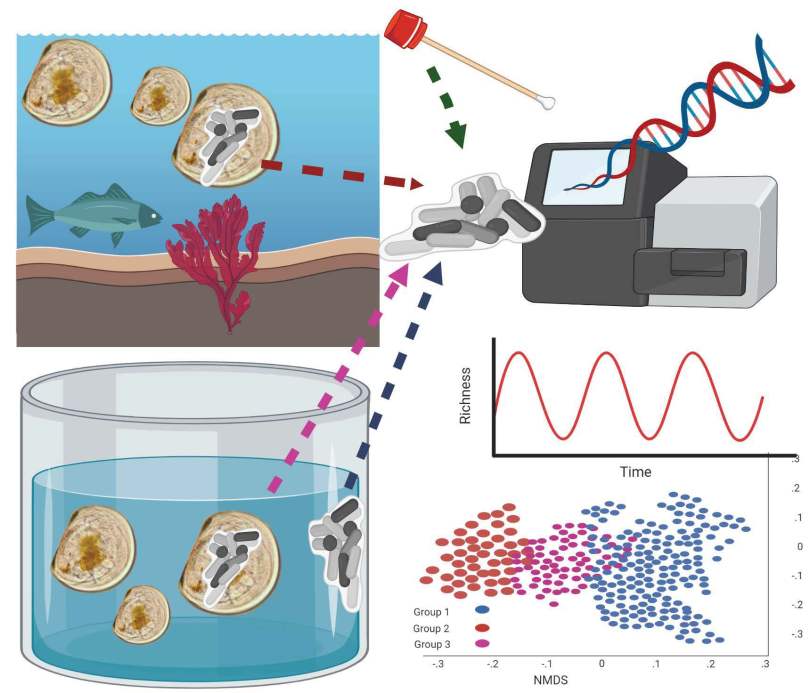
We sampled from some wild larvae, hatchery larvae, and from tank biofilms to indentify what was there. There were two styles of tank setup, and we collected from used tanks as well as tanks after they had been cleaned and refilled with filtered seawater.


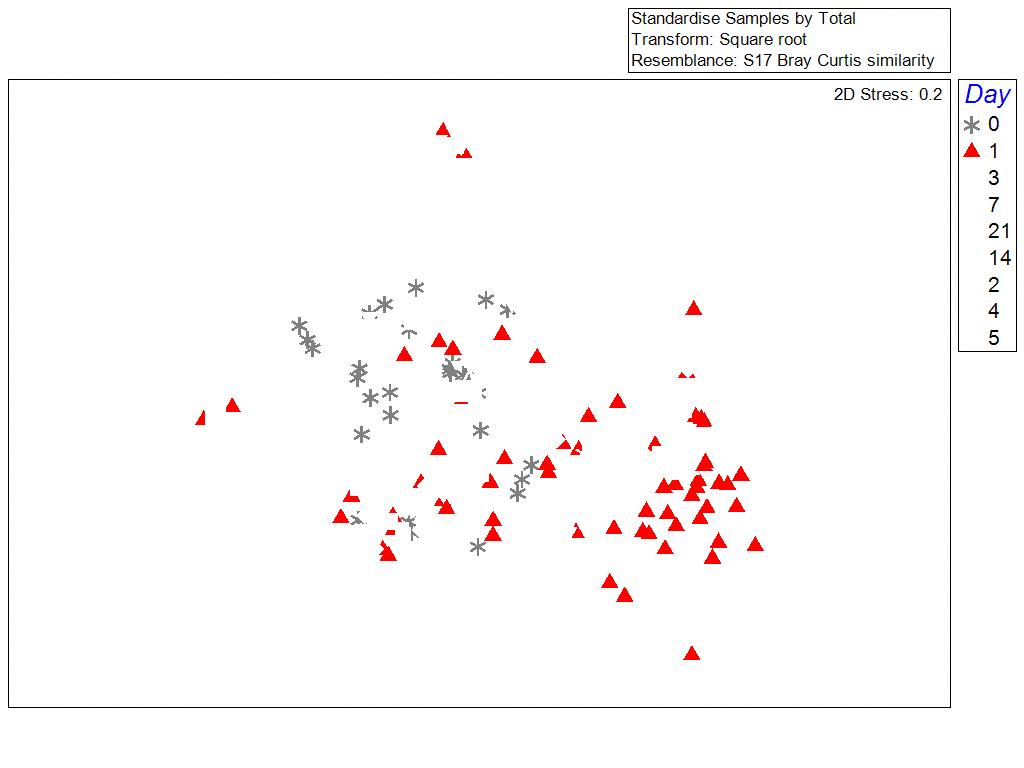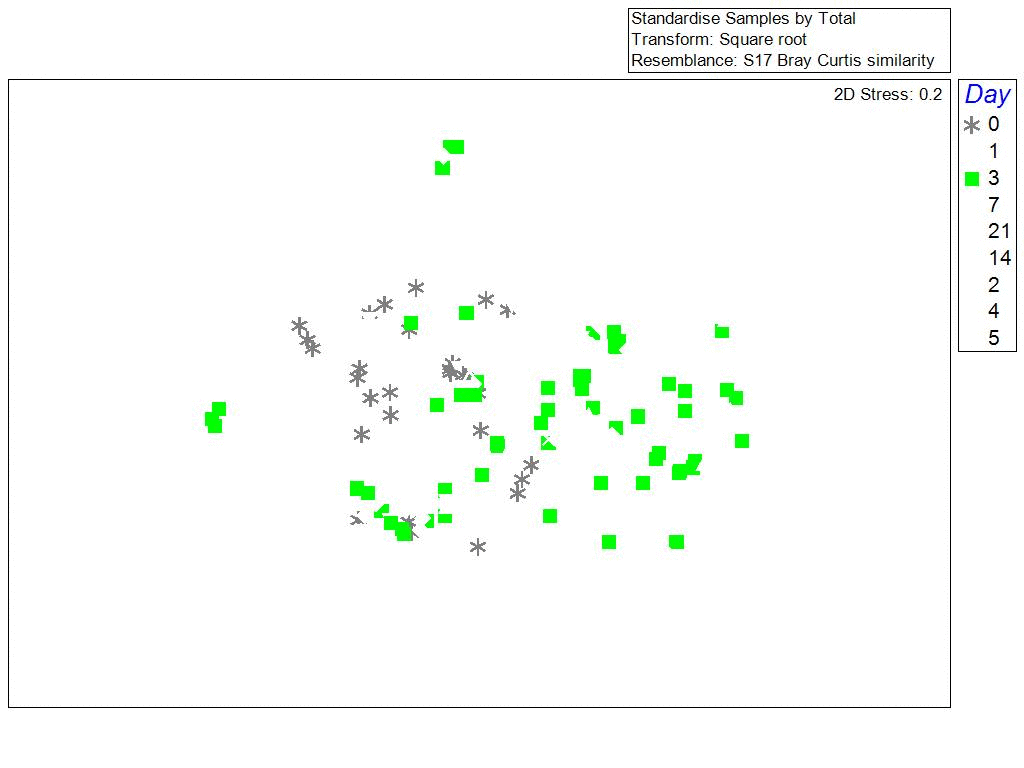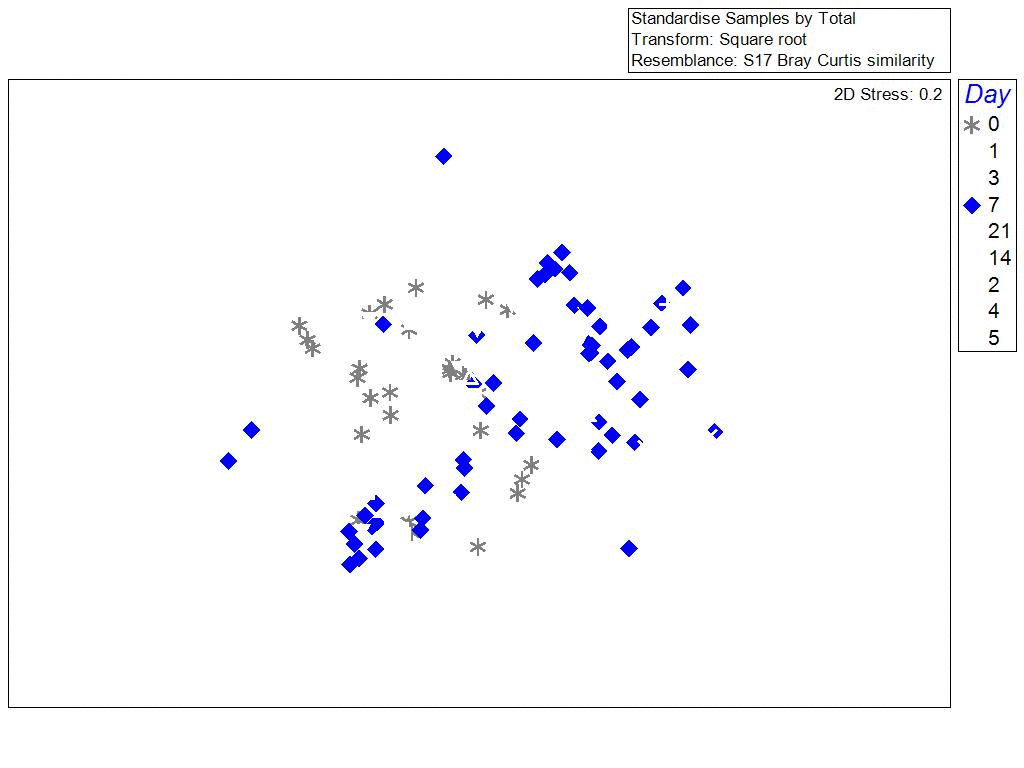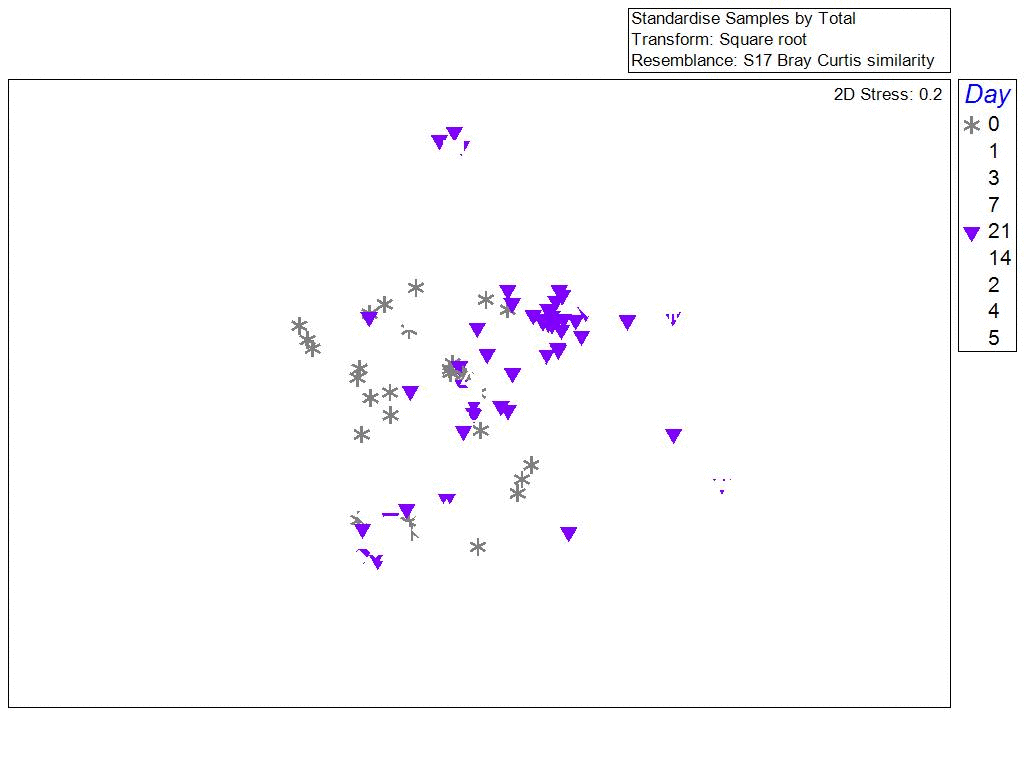
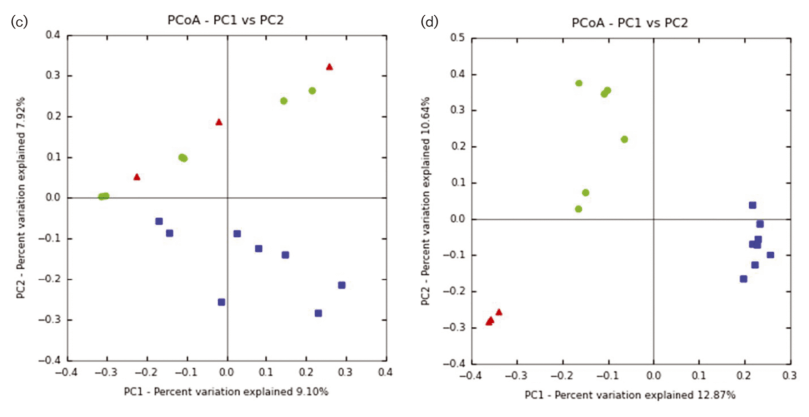
One of the surprising things we found, was that the bacterial communities in biofilms along the sides of larvae tank were more similar to each other (clustering) when samples were collected during the same phase of the lunar cycle. Bacterial richness and community similarity between tank samples fluctuated over the trial in repeated patterns of rise and fall, which showed some correlation to lunar cycle where richness is high when the moon is about 50% and richness is low during new and full moon phases. This may be a proxy for the effects of spring tides and trends in seawater bacteria and phages which are propagated into hatchery tanks. The number of days since the full moon was significantly correlated with bacterial community richness in tanks: low during the full moon, peaking ~ 21 days after the full moon, and decreasing again at the next full moon.

These results along with future work, will inform hatcheries on methods that will increase larval survival in these facilities, for example, implementing additional filtering or avoiding seawater collection during spring tides, to reduce certain bacterial taxa of concern or promoting a more diverse microbial community which would compete against pathogens.
Bacterial community trends associated with sea scallop, Placopecten magellanicus, larvae in a hatchery system.
Authors: Suzanne L. Ishaq1*, Sarah Hosler1, Adwoa Dankwa1, Phoebe Jekielek2, Damian C. Brady3, Erin Grey4,5, Hannah Haskell6, Rachel Lasley-Rasher6, Kyle Pepperman7, Jennifer Perry1, Brian Beal8, Timothy J. Bowden1
Affiliations:1 School of Food & Agriculture, University of Maine, Orono ME 044692 Ecology and Environmental Sciences, University of Maine, Orono ME 044733 School of Marine Sciences, Darling Marine Center, University of Maine, Walpole ME 045734 School of Biology and Ecology, University of Maine, Orono ME 044695 Maine Center for Genetics in the Environment, University of Maine, Orono ME 044696 Department of Biological Sciences, University of Southern Maine, Portland ME 041037 Downeast Institute, Beals, ME 046118 Division of Environmental & Biological Sciences, University of Maine at Machias, Machias, ME 04654
Abstract
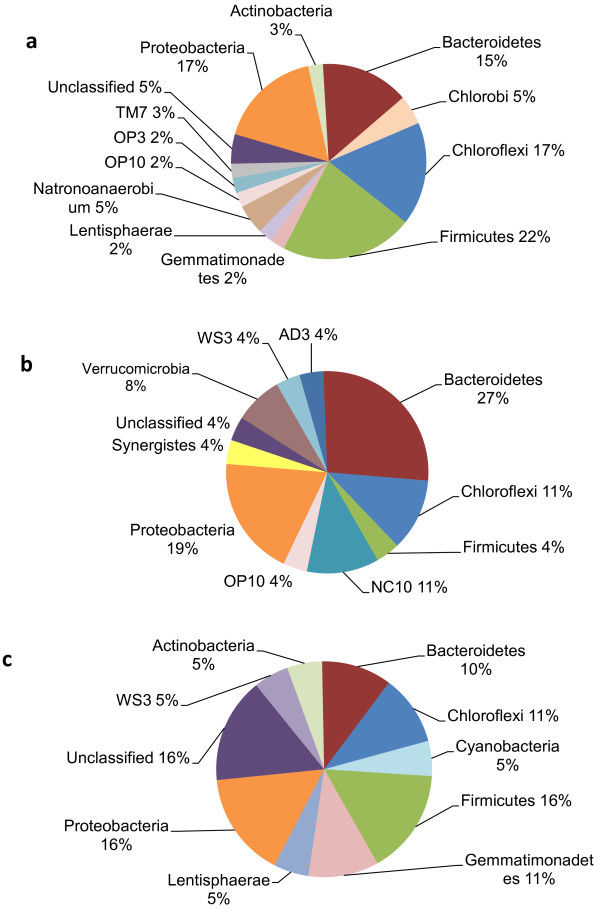
Atlantic sea scallops, Placopecten magellanicus, are the most economically important marine bivalves along the northeastern coast of North America. Wild harvest landings generate hundreds of millions of dollars, and wild-caught adults and juvenile spat are increasingly being cultured in aquaculture facilities and coastal farms. However, the last two weeks of the larval maturation phase in hatcheries are often plagued by large mortality events. Research into other scallop- and aquacultured-species point to bacterial infections or altered functionality of microbial communities which associate with the host. Despite intense filtering and sterilization of seawater, and changing tank water every 48 hours, harmful microbes can still persist in biofilms and mortality is still high. There are no previous studies of the bacterial communities associated with the biofilms growing in scallop hatchery tanks, nor studies with wild or hatchery sea scallops. We characterized the bacterial communities in veliger-stage wild or hatchery larvae, and tank biofilms using the 16S rDNA gene V3-V4 region sequenced on the Illumina MiSeq platform. Hatchery larvae had lower bacterial richness (number of bacteria taxa present) than the wild larvae and tank biofilms, and hatchery larvae had a similar bacterial community (which taxa were present) to both wild larvae and tank biofilms. Bacterial richness and community similarity between tank samples fluctuated over the trial in repeated patterns of rise and fall, which showed some correlation to lunar cycle that may be a proxy for the effects of spring tides and trends in seawater bacteria and phages which are propagated into hatchery tanks. These results along with future work, will inform hatcheries on methods that will increase larval survival in these facilities, for example, implementing additional filtering or avoiding seawater collection during spring tides, to reduce bacterial taxa of concern or promote a more diverse microbial community which would compete against pathogens.
Acknowledgements
The authors would like to thank the staff at the Downeast Institute for supporting the development and implementation of this project, as well as for financially supporting the DNA sequencing; Meredith White of Mook Sea Farm for sharing her expertise and collecting biofilm samples; the Darling Marine Center for sharing their expertise and collecting biofilm samples; and the Sea Scallop Hatchery Implementation (Hit) Team for their expertise, review of this work, and funding support, who are financially supported by the Atlantic States Marine Fisheries Commission and Michael & Alison Bonney. The authors thank Lilian Nowak for assistance with related lab work to this project, and the Map Top Scholars Program for related financial support. The authors also thank Nate Perry for helping us collect wild scallop larvae. All authors have read and approved the final manuscript. This project was supported by the USDA National Institute of Food and Agriculture through the Maine Agricultural & Forest Experiment Station, Hatch Project Numbers: ME0-22102 (Ishaq), ME0-22309 (Bowden), and ME0-21915 (Perry); as well through NSF #OIA-1849227 to Maine EPSCoR at the University of Maine (Grey). This project was supported by an Integrated Research and Extension Grant from the Maine Food and Agriculture Center, with funding from the Maine Economic Improvement Fund.
Ishaq, S.L., Hosler2, S., Dankwa, A., Jekielek, P., Brady, D.C., Grey, E., Haskell, H., Lasley-Rasher, R., Pepperman, K., Perry, J., Beal, B., Bowden, T.J. 2023. Bacterial community trends associated with sea scallop, Placopecten magellanicus, larvae in a hatchery system. Aquaculture Reports 32: 101693.
In the media
“UMaine study reveals how to turn the tide on scallop-rearing challenges”, UMaine News, Feb 5, 2024.