This project expanded upon my work with moose bacteria from three geographic locations, to explore whether there were differences in methanogenic archaea or ciliated protozoa based on location.
Archaea are microorganisms in their own Domain, as they are neither Bacteria nor Eukaryota, although they often have similarities to organisms found in the other two domains. Archaea are found in many extreme environments, but those found in the digestive tract of animals and humans come from the phylum Euryarchaeota. Methanogens require hydrogen to make energy for themselves, and in that process (methanogenesis) methane is created as a byproduct. In the digestive tract, especially in ruminants where the fermentation of plants creates a lot of hydrogen, the presence of methanogens acts a hydrogen sink and can prevent the build up of hydrogen which would otherwise lower the gut pH and be detrimental to both host and microbes. To date, it is unclear if methanogens have any other health effect.
Protozoa are single-celled eukaryotes, and depending on which species they are, can be beneficial or pathogenic. Typically, protozoa in the digestive tract of humans or other monogastrics are pathogens obtained from drinking contaminated water. However, the digestive tracts of monogastrics (ex. humans) and ruminants (ex. moose) are very different, and the later can support a much different microbial community. Specifically, protozoa found in ruminants that have cilia to move around (i.e. ciliated protozoa or ciliates) can have a number of roles, including fermentation of fiber or starch, or predation of bacteria and fungi. As they are so difficult to maintain in culture and study in the lab, the role of protozoa in contributing to host health or methanogenesis is understudied.
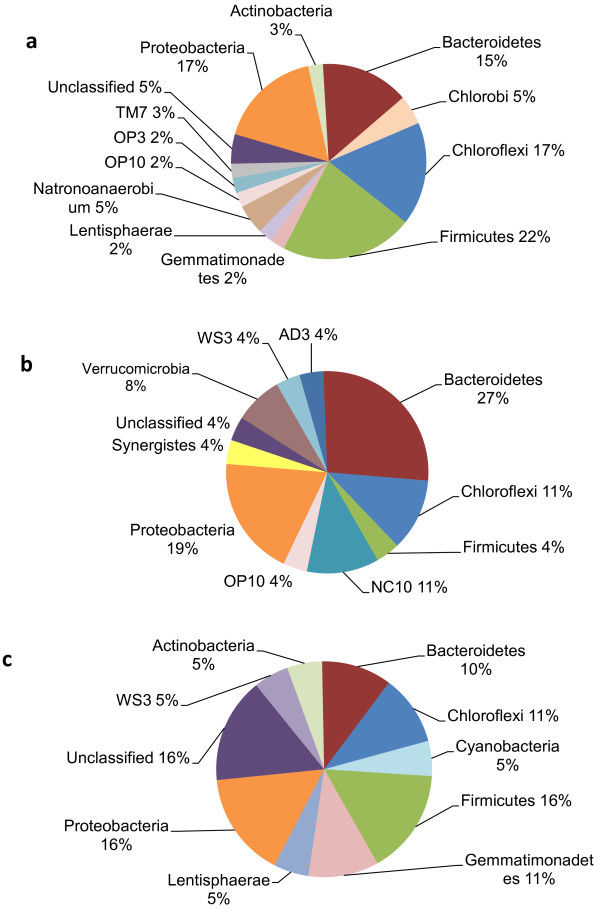
Moose methanogen communities were significantly different between moose in Vermont, Norway, and Alaska, but maintained a core of shared taxa across all populations. This implies that the moose rumen environment (pH, salt content, turnover, host-microbe interactions, etc.) is suitable for only a small number of methanogen species, and that this regulates the community as much as diet might. Methanogen communities were also different based on sex of the moose, and age/weight.

On the other hand, protozoal communities were dramatically different between moose in Vermont, Norway, and Alaska, and shared far fewer taxa. This was surprising, as previous studies on deer had shown a core protozoal community across multiple geographically-separated populations. These moose populations had not been geographically isolated long, but we hypothesized that diet was a much stronger driver of rumen protozoal diversity than previously thought.
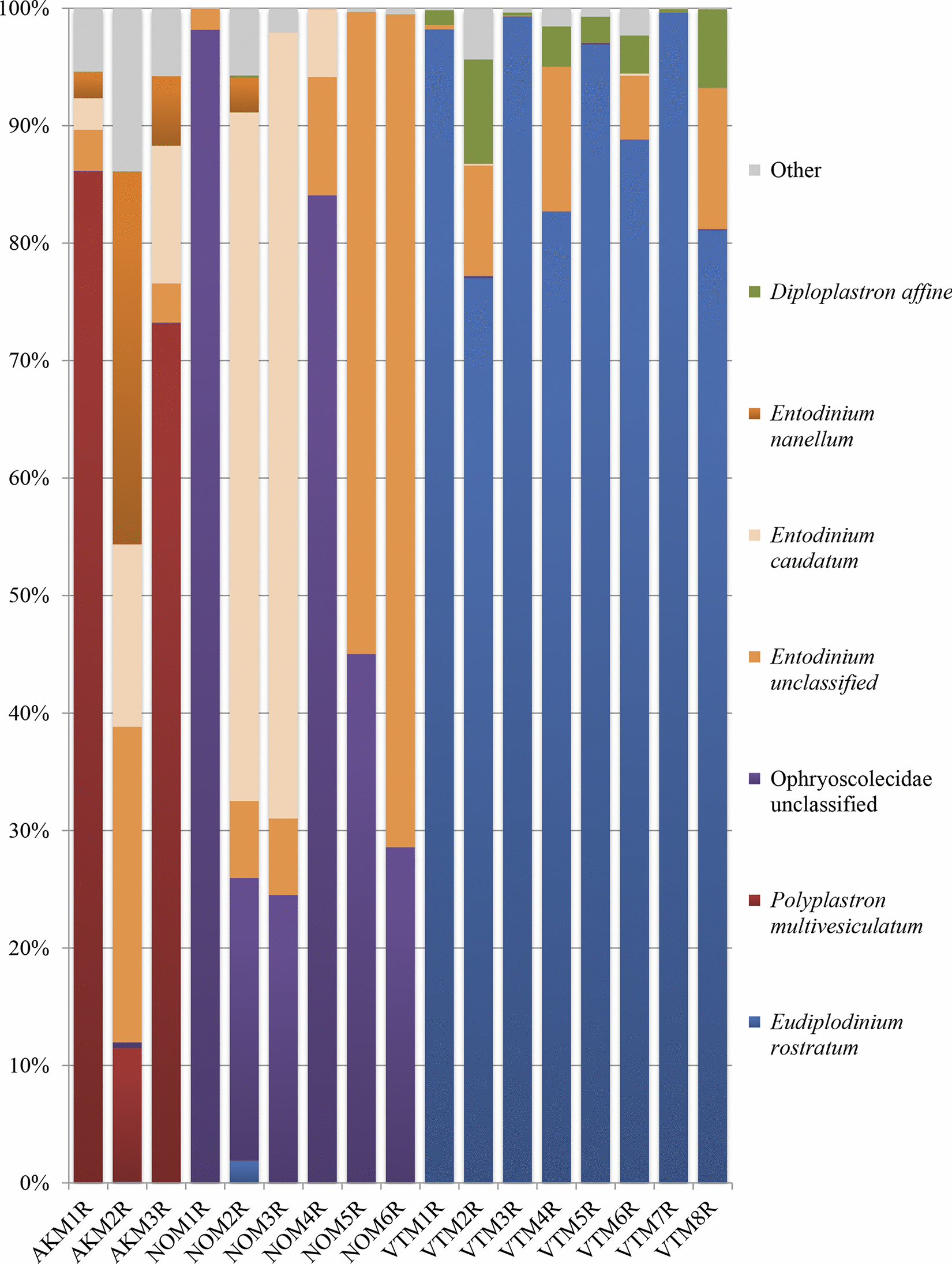
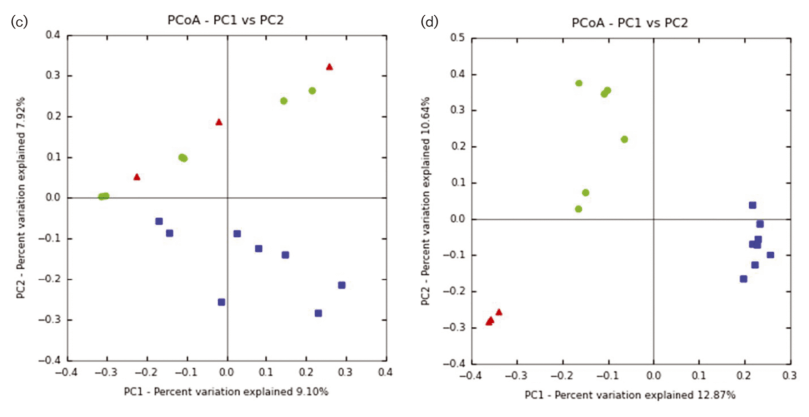
Featured Image; Figure 1: PCoA for moose methanogens (A, C, E) and protozoa (B, D, F). PCoA is coloured by (A, B) gender: female, red; male, blue; (C, D) location: Alaska, red; Norway, green; Vermont, blue; and (E, F) weight class: 1–100 kg, red triangle; 101–200 kg, yellow triangle; 201–300 kg, green down-facing triangle; 301–400 kg, green right-facing triangle, >400 kg (live weight), light blue circle; not available, blue square.
Ishaq, S.L., Sundset, M.A., Crouse, J., Wright, A-D.G. 2015. High-throughput DNA sequencing of the moose rumen from different geographical locations reveals a core ruminal methanogenic archaeal diversity and a differential ciliate protozoal diversity. Microbial Genetics, 2015(1). Article