Summary reposted from the MSE newsletter, now hosted by AMI!Subscribe to the newletter or join the group here.
MSE recently hosted its 5th annual summit, this year dedicated to exploring the concept of Microbiome Stewardship. Microbiome Stewardship is a concept that is intended to provide guiding insights, articulate responsibilities, and suggest practices aimed at maintaining microbial biodiversity and microbiome functioning across microbial habitats, which, in turn, supports the health and well-being of humans, animals, plants, and ecosystems. The summit consisted of two days of presentations (webinars) and two days of virtual workshop discussions.
We opened with two days of webinars featuring 12 speakers from wide-ranging fields of expertise, all focused on how social or environmental conditions impact health and microbiomes. This included an introduction to the concept of microbiome stewardship and guiding principles for its implementation, the need for diversification of fecal microbiome donors for health interventions, degradation of waterways and microbial transfer, the industrialization of food systems and the rise of antimicrobial resistance, the use of too-vague population descriptors in microbiome science, integrating systems-level thinking in microbiology curricula, and working with Indigenous communities on microbiome research. The webinars sparked imaginative and thoughtful questions from the 200 attendees (nearly 300 registrants), and set the stage for the subsequent two days of workshops. We also shared a working draft of the Microbiome Stewardship Guiding Principles document with attendees, and welcomed feedback. We hope to submit that manuscript for peer review and publication soon.
Workshop attendance was by application, and restricted to 50 attendees across the two days, which focused on host and environmental microbiomes, respectively. For each workshop day, attendees self-organized into breakout rooms focusing on different disciplines or themes. Speakers, MSE and Microbiome Stewardship researchers, and attendees discussed the challenges and opportunities for their respective fields, what was needed to achieve more integration between research and education or policy, and how to incorporate the principles of stewardship into their respective research. These conversations helped realize existing areas of overlap between our work, and identify compatible expertise that was needed to explore these interdisciplinary research questions. Similar themes and challenges emerged across workshop days and discussion groups, highlighting opportunities to strengthen the microbiome stewardship and paths to implementation.
The live sessions were recorded to accommodate our global audience who were unable to make the session, and can be viewed here. While the workshops were not recorded, the thoughtful discourse from throughout the seminar and workshops will be used to inform that guiding principles publication-in-development, as well as future publications and output over the next 2-3 years from the collaborations which germinated during the breakout room sessions.
Image Source: Wikimedia Commons, 2017-09-09 Oregon Ducks vs. Nebraska Cornhuskers
The picture is just one instant in an event involving hundreds or thousands of organisms that were all doing a lot of different things, sometimes for just a few seconds. How would you describe it?
Maybe using the number of members present in this community? Or a list of names of attendees? The 16S rRNA gene for prokaryotes, or the 18S rRNA or ITS genes for eukaryotes, for examples, would tell us that. Those genes are found in all types of those organisms, and is a pretty effective means of basic identification. But, it’s only as good as how often that gene is found in the organisms you are looking for. There is no one gene that’s found exactly the same in all organisms, so you might need to target multiple different identification genes to look at all the different types of microorganisms, such as bacteria, fungi, protozoa, or archaea. Viruses don’t share a common gene across types, to look at viruses you’d need something else.
From our identification genes we could identify all the organisms wearing yellow; ex. phylogenetic Family = Ducks. That wouldn’t tell us if they were always found in this ecosystem (native Eugene population) or just passing through (transient population), but we could figure that out if we looked at every home game of the season and found certain community members there time and again.
But knowing they are Ducks doesn’t tell us anything else about that community member. What will they do if it starts raining? Are they able to go mountain biking? Perhaps we could identify their potential for activity by looking at the objects they are carrying? That would be akin to metagenomics, identifying all the DNA present from all the organisms, which tells us what genes are present, but not if they are currently or ever used. It can be challenging to interpret: think of sequencing data from one organism’s genome as one 1,000,000-piece puzzle and all the genomes in a community as 1,000 1,000,000-piece puzzles all dumped in a pile. In the crowd, metagenomics would tell us who had a credit card that was specifically used to buy umbrellas, but not whether they’d actually use the umbrella if it rains (ex. Eugeneans would not).
We could describe what everyone is doing at this moment. That would be transcriptomics, identifying all the RNA to determine which genes were actively being transcribed into proteins for use in some cellular function. If we see someone in the crowd using that credit card for an umbrella (DNA), the receipt would be the RNA. RNA is a working copy you make of the DNA to take to another part of the cell and use as a blueprint to make a protein. You don’t want your entire genome moving around, or need it to make one protein, so you make a small piece of RNA that will only hang around for a short period before degrading (i.e. you crumpling that RNA receipt and throwing it away because who keeps receipts anymore).
Using transcriptomics, we’d see you were activating your money to get that umbrella, but we wouldn’t see the umbrella itself. For that, we’d need metabolomics, which uses chemistry and physics instead of genomics, in order to identify chemicals (most often proteins). Think of metabolomics as describing this crowd by all the trash and crumbs and miscellaneous items they left behind. It’s one way to know what biological processes occurred (popcorn consumption and digestion).
Image Source: Wikimedia Commons, Metabolomics
From a technical standpoint, researching a microbiome might mean looking at all the DNA from all the organisms present to know who they are and of what they are capable. It might also mean looking at all the RNA present, which would tell you what genes were being used by “everyone” for whatever they were doing at a particular moment. Or you might also add metabolomics to identify all the chemical metabolites, which would be all the end products of what those cells were doing, and which are more stable than RNA so they could give you data about a longer frame of time. Collectively, -omics are technology that looks at all of a certain biological substance to help you understand a dynamic community. However, it’s important to remember that each technology gives a particular view of the community and comes with its own limitations.
To study DNA or RNA, there are a number of “wet-lab” (laboratory) and “dry-lab” (analysis) steps which are required to access the genetic code from inside cells, polish it to a high-sheen such that the delicate technology we rely on can use it, and then make sense of it all. Destructive enzymes must be removed, one strand of DNA must be turned into millions of strands so that collectively they create a measurable signal for sequencing, and contamination must be removed. Yet, what constitutes contamination, and when or how to deal with it, remains an actively debated topic in science. Major contamination sources include human handlers, non-sterile laboratory materials, other samples during processing, and artificial generation due to technological quirks.
Contamination from human handlers
This one is easiest to understand; we constantly shed microorganisms and our own cells and these aerosolized cells may fall into samples during collection or processing. This might be of minimal concern working with feces, where the sheer number of microbial cells in a single teaspoon swamp the number that you might have shed into it, or it may be of vital concern when investigating house dust which not only has comparatively few cells and little diversity, but is also expected to have a large amount of human-associated microorganisms present. To combat this, researchers wear personal protective equipment (PPE) which protects you from your samples and your samples from you, and work in biosafety cabinets which use laminar air flow to prevent your microbial cloud from floating onto your workstation and samples.
Fun fact, many photos in laboratories are staged, including this one, of me as a grad student. I’m just pretending to work. Reflective surfaces, lighting, cramped spaces, busy scenes, and difficulty in positioning oneself makes “action shots” difficult. That’s why many lab photos are staged, and often lack PPE.
Photo Credit: Kristina Drobny
Contamination from laboratory materials
Microbiology or molecular biology laboratory materials are sterilized before and between uses, perhaps using chemicals (ex. 70% ethanol), an ultraviolet lamp, or autoclaving which combines heat and pressure to destroy, and which can be used to sterilize liquids, biological material, clothing, metal, some plastics, etc. However, microorganisms can be tough – really tough, and can sometimes survive the harsh cleaning protocols we use. Or, their DNA can survive, and get picked up by sequencing techniques that don’t discriminate between live and dead cellular DNA.
In addition to careful adherence to protocols, some of this biologically-sourced contamination can be handled in analysis. A survey of human cell RNA sequence libraries found widespread contamination by bacterial RNA, which was attributed to environmental contamination. The paper includes an interesting discussion on how to correct this bioinformatically, as well as a perspective on contamination. Likewise, you can simply remove sequences belonging to certain taxa during quality control steps in sequence processing. There are a number of hardy bacteria that have been commonly found in laboratory reagents and are considered contaminants, the trouble is that many of these are also found in the environment, and in certain cases may be real community members. Should one throw the Bradyrhizobium out with the laboratory water bath?
Chimeras
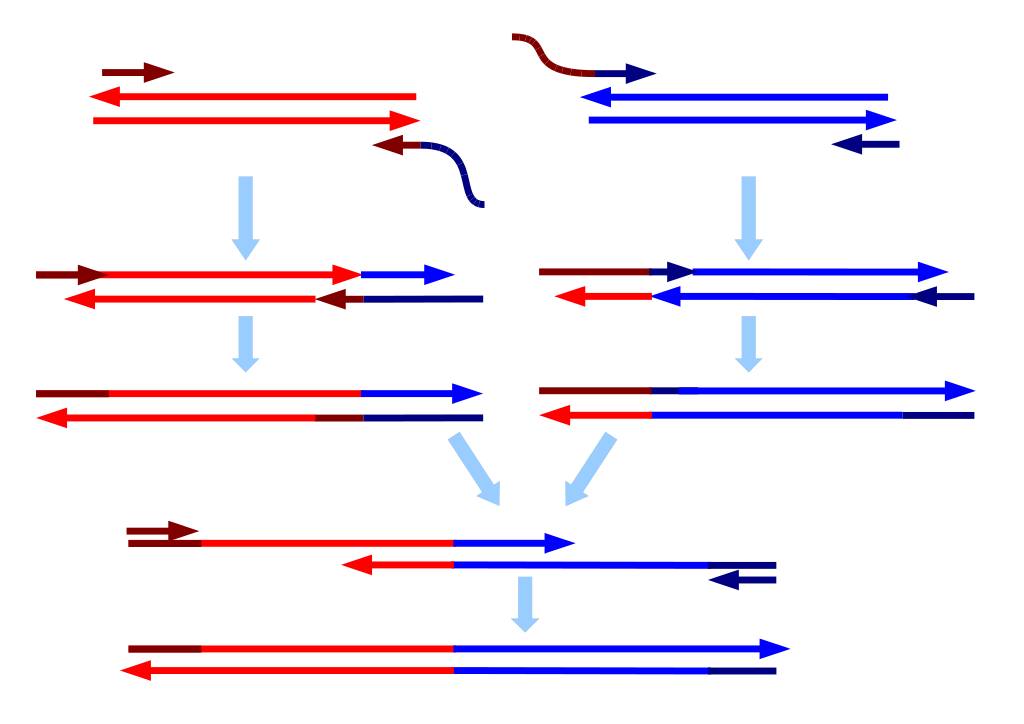
Like the mythical creatures these are named for, sequence chimeras are DNA (or cDNA) strands which are accidentally created when two other DNA strands merged. Chimeric sequences can be made up of more than two DNA strand parents, but the probability of that is much lower. Chimeras occur during PCR, which takes one strand of genetic code and makes thousands to millions of copies, and a process used in nearly all sequencing workflows at some point. If there is an uneven voltage supplied to the machine, the amplification process can hiccup, producing partial DNA strands which can concatenate and produce a new strand, which might be confused for a new species. These can be removed during analysis by comparing the first and second half of each of your sequences to a reference database of sequences. If each half matches to a different “parent”, it is deemed chimeric and removed.
During DNA or RNA extraction, genetic code can be flicked from one sample to another during any number of wash or shaking steps, or if droplets are flicked from fast moving pipettes. This can be mitigated by properly sealing all sample containers or plates, moving slowly and carefully controlling your technique, or using precision robots which have been programmed with exacting detail — down to the curvature of the tube used, the amount and viscosity of the liquid, and how fast you want to pipette to move, so that the computer can calculate the pressure needed to perform each task. Sequencing machines are extremely expensive, and many labs are moving towards shared facilities or third-party service providers, both of which may use proprietary protocols. This makes it more difficult to track possible contamination, as was the case in a recent study using RNA; the researchers found that much of the sample-sample contamination occurred at the facility or in shipping, and that this negatively affected their ability to properly analyze trends in the data.
Sample-sample contamination during sequencing
Controlling sample-sample contamination during sequencing, however, is much more difficult to control. Each sequencing technology was designed with a different research goal in mind, for example, some generate an immense amount of short reads to get high resolution on specific areas, while others aim to get the longest continuous piece of DNA sequenced as possible before the reaction fails or become unreliable. they each come with their own quirks and potential for quality control failures.
Due to the high cost of sequencing, and the practicality that most microbiome studies don’t require more than 10,000 reads per sample, it is very common to pool samples during a run. During wet-lab processing to prepare your biological samples into a “sequencing library”, a unique piece of artificial “DNA” called a barcode, tag, or index, is added to all the pieces of genetic code in a single sample (in reality, this is not DNA but a single strand of nucleotides without any of DNA’s bells and whistles). Each of your samples gets a different barcode, and then all your samples can be mixed together in a “pool”. After sequencing the pool, your computer program can sort the sequences back into their respective samples using those barcodes.
While this technique has made sequencing significantly cheaper, it adds other complications. For example, Illumina MiSeq machines generate a certain number of sequence reads (about 200 million right now) which are divided up among the samples in that run (like a pie). The samples are added to a sequencing plate or flow cell (for things like Illumina MiSeq). The flow cells have multiple lanes where samples can be added; if you add a smaller number of samples to each lane, the machine will generate more sequences per sample, and if you add a larger number of samples, each one has fewer sequences at the end of the run. you have contamination. One drawback to this is that positive controls always sequence really well, much better than your low-biomass biological samples, which can mean that your samples do not generate many sequences during a run or means that tag switching is encouraged from your high-biomass samples to your low-biomass samples.
Cross-contamination can happen on a flow cell when the sample pool wasn’t thoroughly cleaned of adapters or primers, and there are great explanations of this here and here. To generate many copies of genetic code from a single strand, you mimic DNA replication in the lab by providing all the basic ingredients (process described here). To do that, you need to add a primer (just like with painting) which can attach to your sample DNA at a specific site and act as scaffolding for your enzyme to attach to the sample DNA and start adding bases to form a complimentary strand. Adapters are just primers with barcodes and the sequencing primer already attached. Primers and adapters are small strands, roughly 10 to 50 nucleotides long, and are much shorter than your DNA of interest, which is generally 100 to 1000 nucleotides long. There are a number of methods to remove them, but if they hang around and make it to the sequencing run, they can be incorporated incorrectly and make it seem like a sequence belongs to a different sample.
This may sound easy to fix, but sequencing library preparation already goes through a lot of stringent cleaning procedures to remove everything but the DNA (or RNA) strands you want to work with. It’s so stringent, that the problem of barcode swapping, also known as tag switching or index hopping, was not immediately apparent. Even when it is noted, it typically affects a small number of the total sequences. This may not be an issue, if you are working with rumen samples and are only interested in sequences which represent >1% of your total abundance. But it can really be an issue in low biomass samples, such as air or dust, particularly in hospitals or clean rooms. If you were trying to determine whether healthy adults were carrying but not infected by the pathogen C. difficile in their GI tract, you would be very interested in the presence of even one C. difficile sequence and would want to be extremely sure of which sample it came from. Tag switching can be made worse by combining samples from very different sample types or genetic code targets on the same run.
There are a number of articles proposing methods of dealing with tag switching using double tags to reduce confusion or other primer design techniques, computational correction or variance stabilization of the sequence data, identification and removal of contaminant sequences, or utilizing synthetic mock controls. Mock controls are microbial communities which have been created in the lab by mixed a few dozen microbial cultures together, and are used as a positive control to ensure your procedures are working. because you are adding the cells to the sample yourself, you can control the relative concentrations of each species which can act as a standard to estimate the number of cells that might be in your biological samples. Synthetic mock controls don’t use real organisms, they instead use synthetically created DNA to act as artificial “organisms”. If you find these in a biological sample, you know you have contamination. One drawback to this is that positive controls always sequence really well, much better than your low-biomass biological samples, which can mean that your samples do not generate many sequences during a run or means that tag switching is encouraged from your high-biomass samples to your low-biomass samples.
Incorrect base calls
Cross-contamination during sequencing can also be a solely bioinformatic problem – since many of the barcodes are only a few nucleotides (10 or 12 being the most commonly used), if the computer misinterprets the bases it thinks was just added, it can interpret the barcode as being a different one and attribute that sequence to being from a different sample than it was. This may not be a problem if there aren’t many incorrect sequences generated and it falls below the threshold of what is “important because it is abundant”, but again, it can be a problem if you are looking for the presence of perhaps just a few hundred cells.
Implications
When researching environments that have very low biomass, such as air, dust, and hospital or cleanroom surfaces, there are very few microbial cells to begin with. Adding even a few dozen or several hundred cells can make a dramatic impactinto what that microbial community looks like, and can confound findings.
Collectively, contamination issues can lead to batch effects, where all the samples that were processed together have similar contamination. This can be confused with an actual treatment effect if you aren’t careful in how you process your samples. For example, if all your samples from timepoint 1 were extracted, amplified, and sequenced together, and all your samples from timepoint 2 were extracted, amplified, and sequenced together later, you might find that timepoint 1 and 2 have significantly different bacterial communities. If this was because a large number of low-abundance species were responsible for that change, you wouldn’t really know if that was because the community had changed subtly or if it was because of the collective effect of low-level contamination.
Stay tuned for a piece on batch effects in sequencing!
In 2016, I was a post-doc in the Menalled Lab, which studies plant and weed ecology in the context of agricultural production and sustainability. There, I assessed soil bacterial communities under different farming management practices and climate scenarios. I also helped to develop a grant proposal, which was just accepted by the USDA AFRI NIFA Agricultural Production Systems! Leading this project is Dr. Fabian Menalled (as Principal Investigator, or PI), along with a number of other PIs; Dr. Amy Trowbridge, Dr. David Weaver, Dr. Tim Seipel, Dr. Maryse Bourgault, and Dr. Carl Yeoman, and collaborators Dr. Darrin Boss, Dr. Kate Fuller, Dr. Ylva Lekberg, and myself as a subaward PI. I will again be providing microbial community analysis for this project, and collectively the project investigators will bring expertise in plant ecology, agronomy, economics, soil and plant chemistry, microbial ecology, agroecosystems, and more.
This research and extension project focuses on the needs of dryland agricultural stakeholders and it was designed in close collaboration with the NARC Advisory Board. While I was only able to attend one meeting, other team members regularly meet with Montana producers to discuss current issues and identify locally-sourced needs for agricultural research. During this project, we will continue to meet with the NARC Advisory Board to share our results, evaluate implications, and better serve the producer community.
Diversifying cropping systems through cover crops and targeted grazing: impacts on plant-microbe-insect interactions, yield and economic returns.
Project summary
The semi-arid section of the Northern Great Plains is one of the
largest expanses of small grain agriculture and low-intensity livestock
production. However, extreme landscape simplification, excessive reliance on
off-farms inputs, and warmer and drier conditions hinder its agricultural
sustainability. This project evaluates the potential of diversifying this region
through the integration of cover crops and targeted grazing. We will complement
field and greenhouse studies to appraise the impact of system diversity,
temperature, and precipitation on key multi-trophic interactions, yields, and
economic outputs. Specifically, we will 1) Assess ecological drivers as well as
agronomic and economic consequences of integrating cover crops and livestock
grazing in semi-arid systems, 2) Evaluate how climate variability modify the
impacts of cover crops and livestock grazing on agricultural outputs. Specifically,
we will 2.1) Compare the effect of increased temperature and reduced moisture
on agronomic and economic performance of simplified and diversified systems,
2.2.) Assess the impact of climate and system diversity on associated biodiversity
(weeds, insect, and soil microbial communities) and above- and belowground
volatile organic (VOC) compound emissions, and 2.3) Evaluate how changes in
microbially induced VOCs influence multitrophic plant-insect interactions.
Objectives
Assess key ecological drivers as well as agronomic and economic consequences of integrating cover crops and livestock grazing in semi-arid production systems
Compare the agronomic and economic performance of simplified and diversified systems
Assess the impact of cover crops and livestock grazing on the associated biodiversity (weeds, insects, and the soil microbiota)
Evaluate how climate conditions modify the impacts of cover crops and livestock grazing on semi-arid production systems
Compare the effect of temperature and soil moisture on agronomic and economic performance of simplified and diversified systems
Assess the impact of climate and system diversity on associated biodiversity and above- and belowground volatile organic compound (VOC) emissions
Evaluate how changes in VOCs emissions influence important multitrophic interactions such as resistance to wheat stem sawfly and natural enemy host location cues
Integrate the knowledge generated into an outreach program aimed at improving producers’ adoption of sustainable diversified crop-livestock systems
For the past two months, I’ve been spending quite a bit of time writing grant proposals. In particular; those which expand our understanding of indoor lighting on human health and behavior, the indoor microbiome, and energy usage in buildings. These project proposals are collaborative efforts between several University of Oregon research labs: Biology and the Built Environment Center, Energy Studies and Buildings Laboratory, and the Baker Lighting Lab. I’ll have more updates in the next few months as those are reviewed.
Siobhan “Shevy” Rockcastle, Chair of the Baker Lighting Lab, and I have been brainstorming ideas, and today I went over to the Baker Lab to check it out in person. The Lab is decorated with concept-design lighting projects from previous students, which are not only beautiful, but extremely creative. Here are a few of my favorites!