I’m delighted to announce a new publication on the importance of preserving microbiomes to secure health in degrading ecosystems! The paper outlines strategies for preserving critical microbes, functions, and microbial communities using some specific examples, and ties these back to opportunities and challenges to making conservation efforts.
This paper was a collaborative effort by several members of the Microbiome Stewardship team, Panuya Athithan (grad student working with Emma), Kieran O’Doherty (fearless leader of the MiSt group), Emma Allen-Vercoe (maven of the human microbiome), and myself. Panuya and I led the paper, weaving our favorite stories of microbial symbioses together with existing studies that support the need for stewardship. Panuya is currently a PhD student working with Emma on a variety of projects, including a gut microbiome and early life project she was interviewed about here. She’s also a Young Director at the non-profit Fora: Network for Change, and was previously an undergraduate researcher while at the University of Waterloo.
A little over a year ago, the author team was discussing the need for papers which outline examples of critical host-microbial or ecosystem-microbial partnerships which are irreplaceable (unless you have several million years of free time to wait for evolution), as a means of supporting calls for taking action now to preserve life and ecosystems on what is currently the only planet we call home.

Over a series of conversations with the MiSt group, as well as during the first public meeting to create the IUCN Microbe Specialist Group, our author team honed our paper to address the concerns of researchers over the ability and practicality of stewardship microbes.
Left to right in the photo are some of the MiSt group; front: Zhongzhi (Michael) Sun, Emma Allen-Vercoe, Sue Ishaq; middle: Mikaela Beijbom, Mallory Choudoir, Sarah Elton, Kieran O’Doherty, Panuya Athithan; back: Grace Gabber, Andreas Heyland, Rob Beiko.
This paper is one of the first in a forthcoming special issue (announcement coming soon!), which will feature several invited papers from my microbiome stewardship colleagues (both original team and expansion pack researchers). These papers will expand upon the concept of what it means to share microbes between individuals, communities, and ecosystems; what it would mean to consider microbes as shared natural resources to which everyone had an innate right to; and how it would look for public and planetary health to reduce the harm of human industry and consumerism to live more sustainably and regain all the benefits that the microbial world can provide us.
Microbes first into the life rafts: preserving microbiomes to secure health in degrading ecosystems
Authors: Panuya Athithan 1,2*, Suzanne L. Ishaq 2,3,4*, Emma Allen-Vercoe 1,2 Kieran C. O’Doherty 2,4,5
- 1 Department of Molecular and Cellular Biology, University of Guelph, Guelph, Ontario, N1G 2W1
- 2 The Microbiome Stewardship research group
- 3 School of Food and Agriculture, University of Maine, Orono, Maine, 04469
- 4 The Microbes and Social Equity working group, Orono, Maine, 04469
- 5 Department of Psychology, University of Guelph, Guelph, Ontario, N1G 2W1
Abstract
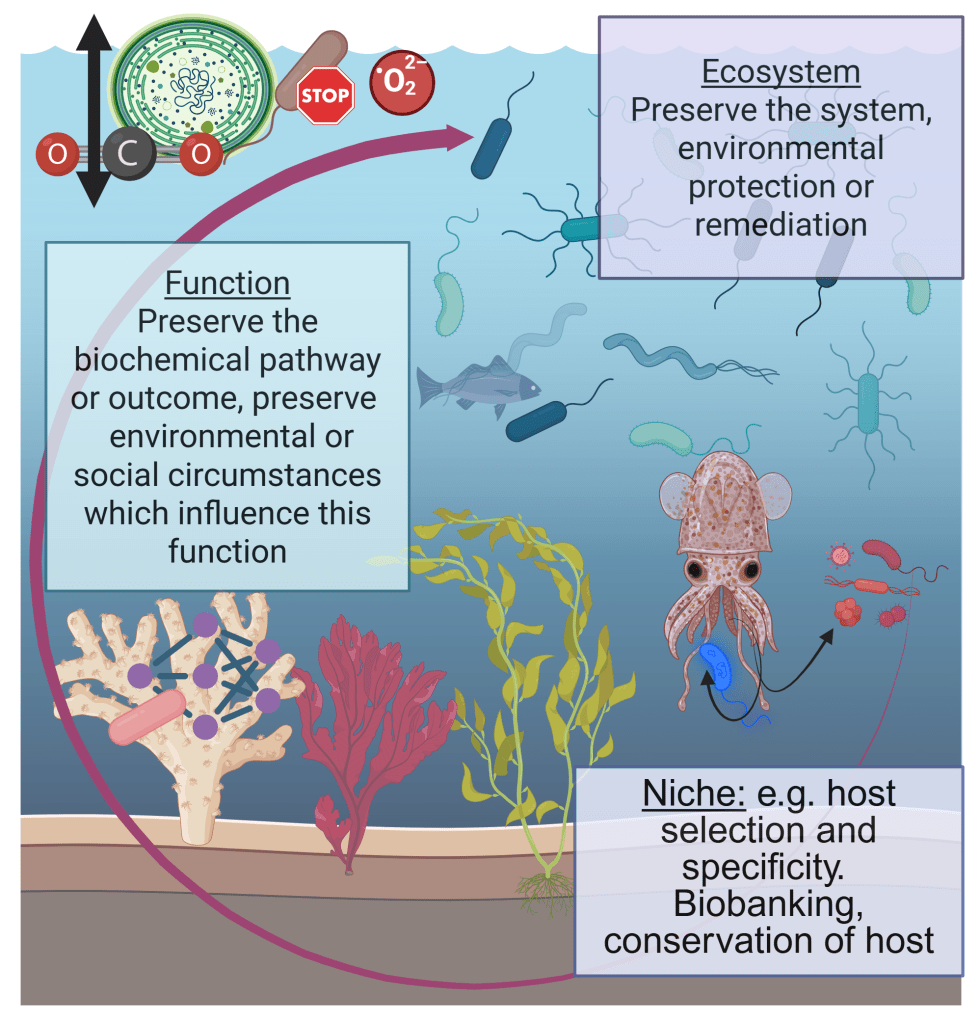
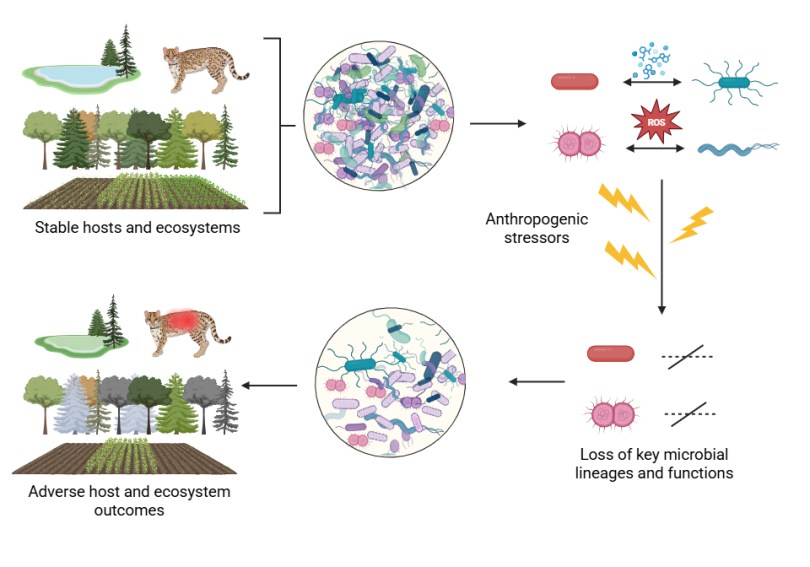
All organisms on the planet intrinsically rely on microbial ecosystems, and there are increasing calls from research communities to consider microbiota when administering personal or public health, ecosystem health, and the use of microbiota in personal or environmental health remediation, such as reducing the impacts of climate change, or protecting at-risk habitats which host rare microbiota. Through our collective work on the integral nature of microbiomes to host and environmental health, on health policy, and on the development of research and policy agendas, we have previously developed the concept of ‘microbiome stewardship’ and guidelines to promote consideration of microbial communities broadly or in specific scenarios. The practicality of stewarding one versus many microbiota is highly contextual, and will require different strategies for different scales of conservation. Here, we provide scientific arguments for the need for microbial stewardship, examples of possible solutions scaled to different ecological challenges or conservation goals, discourse on the logistical challenges which have been cited by research communities, and opportunities to use cutting-edge microbiome concepts and technology to implement large-scale interventions.
Sustainability Statement
Microorganisms are responsible for environmental and organismal health, and the stewardship of microbiota has applications for human, plant, animal, and environmental health on local and global scales. The concepts described here are pertinent, in particular, to the United Nations’ Sustainable Development Goal 3) good health and wellbeing. Additionally, our paper is relevant to goals 6) clean water and sanitation, 9) industry, innovation, and infrastructure, 11) sustainable cities and communities, 12) responsible consumption and production, 13) climate action, 14) life below water, 15) life on land, and 17) partnerships for the goals.